

p (1).png)



.png)




.png)

Research Article
Development and Characterization of Self-Emulsifying Drug Delivery System (Sedds) for a Bcs Class Ii Drug
- Dr. Ashish Sarkar
Corresponding author: Dr. Ashish Sarkar, Dean School of Pharmacy YBN University Ranchi- Jharkhand-834010.
Volume: 2
Issue: 6
Article Information
Article Type : Research Article
Citation : Abhigyan Wenkateswaraun, Jayashri Behera, Dr. Ashish Sarkar. Development And Characterization of Self-Emulsifying Drug Delivery System (Sedds) for a Bcs Class Ii Drug. Journal of Medical and Clinical Case Reports 2(6). https://doi.org/10.61615/JMCCR/2025/OCT027141010
Copyright: © 2025 Dr. Ashish Sarkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI: https://doi.org/10.61615/JMCCR/2025/OCT027141010
Publication History
Received Date
19 Sep ,2025
Accepted Date
04 Oct ,2025
Published Date
10 Oct ,2025
Abstract
Oral delivery presents unique challenges, as the bioavailability of many BCS Class II drugs is low and difficult to estimate because of their persistent weak solubility in water. In order to overcome this limitation, research and development of a self-emulsifying drug delivery system (SEDDS) were carried out. Since valsartan is a commonly used drug for hypertension, it was selected as a model medicine for the BCS Class II category. Even though its absorption is hindered in the acidic environment of the esophagus and stomach, its oral bioavailability is only around 25%. The reason for this is that it has a low rate of environmental absorption. The chemical composition of the mixture was determined to include isopropyl myristate in the oil phase, Capryol 90 and Tween 20 as surfactants, and Transcutol HP as a cosurfactant. The results of the solubility tests confirmed this. The component ratios were optimized using pseudo-ternary phase diagrams. Wet granulation, Avicel pH101, and hydroxypropyl methylcellulose (HPMC) were used to solidify the liquid SEDDS formulations. The intended outcomes necessitated this action. The formulations were tested for efficiency in emulsification, droplet size, and in vitro drug release. When tested in conditions meant to mimic the stomach (pH 1.2), the SEDDS and solid-SEDDS systems substantially increased valsartan's solubility. The commercial formulation had a much lower solubility before this. The release of the Avicel-based solid SEDDS was shown to be pH-insensitive and unaffected by the patient's dietary status or rapid eating, or fasting. These findings suggest that SEDDS is a strategy worth investigating further as a means to circumvent the solubility limitations of BCS Class II drugs and increase their oral bioavailability.
Keywords: Self-emulsifying drug delivery system (SEDDS), Solid SEDDS (S-SEDDS), Valsartan, BCS Class II drug, Solubility enhancement, Bioavailability improvement
Development and Characterization of Self-Emulsifying Drug Delivery System (Sedds) for a Bcs Class Ii Drug
Abhigyan Wenkateswaraun1, Jayashri Behera2, Dr. Ashish Sarkar3*
1PG Scholar School of Pharmacy YBN University.
2Associate Professor, School of Pharmacy, YBN University.
3Dean School of Pharmacy YBN University Ranchi, Jharkhand-834010.
Introduction
It is estimated that around 17.9 million people pass away each year due to cardiovascular diseases (CVDs), making them the leading cause of premature mortality worldwide [1]. Hypertension is the most important risk factor that can be altered, and it is the major cause of cardiovascular morbidity [2]. This includes heart failure, myocardial infarction, and stroke are all examples of cardiovascular morbidity. There is a rapidly increasing prevalence of hypertension, which is driven by a number of causes, including an aging population, changes in eating choices, and sedentary lifestyles [3]. This is a significant public health problem that needs an immediate response. It may still be difficult to get optimal treatment outcomes due to challenges with drug solubility and bioavailability of certain antihypertensive medicines, despite the fact that medication has significantly reduced the number of problems associated with hypertension.
Biopharmaceutics Classification System (BCS) and Challenges with Class II Drugs
The Biopharmaceutics Classification System (BCS) was developed to categorize drugs according to their solubility and intestinal permeability [4]. This was done in order to improve researchers' ability to predict how pharmaceuticals would be absorbed into the circulation after administration. Based on this method, the absorption rate of BCS Class II medicines is most strongly correlated with the dissolution phase. This is due to the fact that these compounds have a low water solubility but a high permeability [5]. Bioavailability is insufficient and fluctuates; the dose must be administered often, and patient compliance is hampered [6]. This is all due to the fact that the drug is poorly soluble in the gastrointestinal tract. There are many hypertension drugs that are included in this category. Valsartan, candesartan, and irbesartan are all examples of these medications; in order to make them more effective when taken orally, different formulation strategies are necessary.
Valsartan as a Model BCS Class II Drug
The treatment of hypertension, heart failure, and post-myocardial infarction is one of the most common reasons for taking valsartan, a selective angiotensin II type 1 (AT1) receptor blocker (ARB) [7]. One mechanism by which valsartan lowers blood pressure is via inhibiting vasoconstriction and aldosterone production through the inhibition of AT1 receptors [8]. This is how valsartan achieves its blood pressure-lowering effects. Despite the fact that valsartan is both safe and effective in clinical settings, it has a pH-dependent solubility problem, which means that it dissolves more readily at a basic pH than it does in the acidic environment of the stomach [9]. As a consequence of this, its oral bioavailability is limited, ranging from 23 and 25 percent, which results in a broad variety of therapeutic responses [10]. It is necessary to find solutions to these solubility-related problems in order to achieve a prolonged antihypertensive effect.
Self-Emulsifying Drug Delivery Systems (SEDDS): A Novel Approach
Recent years have seen a surge in interest in lipid-based formulations such as self-emulsifying drug delivery systems (SEDDS) as a means to increase the oral absorption and solubility of substances that are not particularly water-soluble [11]. The need to enhance the efficacy of these formulations has stoked this interest. Gently combining SEDDS—isotropic mixtures of oils, surfactants, and co-surfactants—with gastrointestinal fluids causes them to sudsify spontaneously [12]. Because of this, while the process is running, nanoemulsions or fine oil-in-water emulsions will be created. This method increases the drug's surface area and enhances its wetting and dissolving characteristics simultaneously. Using SEDDS may also have the added advantage of increasing systemic exposure and decreasing first-pass metabolism. Medications may now be more easily absorbed by the body via the intestinal lymphatic system as a result of these advantages [13]. The use of adsorbents as Avicel pH101, Aerosil, or hydroxypropyl methylcellulose (HPMC), to transform liquid SEDDS into solid dosage forms (S-SEDDS) is a major step forward in this field [14]. Controlled release profiles are one of several benefits made possible by this solidification. The benefits of solidification include improved patient compliance, mobility, and stability.
Valsartan is an effective and well-tolerated hypertension medicine; however, it is not very helpful as a treatment since it is poorly soluble in water and has low oral bioavailability. Evidence from previous studies suggests that lipid-based drug delivery methods may significantly improve the bioavailability and solubility of BCS Class II medications [15-16]. Therefore, this study aims to create and describe a valsartan self-emulsifying drug delivery system (SEDDS), with a focus on excipient optimization, pseudo-ternary phase diagram development, and in vitro assay efficiency assessment. Moreover, the study investigates the feasibility of solidifying liquid SEDDS to provide pH-independent solubility and continuous release. In theory, this might be a much better alternative to the current processes used to make valsartan.
Objectives
- To create and improve a SEDDS in order to improve valsartan's solubility and dissolution.
- To describe droplet size, emulsification, drug release, and pH-independent solubility of the produced SEDDS and solid-SEDDS
Method
Screening Tests
The shake-flask method was used in order to ascertain the manner in which VST dissolves in a variety of oils, surfactants, and co-surfactants. In a nutshell, two milliliters of each component were combined with additional VST, and then the mixture was allowed to stir in a shaker that moved at a speed of two hundred revolutions per minute (CAT S20, Germany) for a period of seventy-two hours. In a centrifuge (Nuve 800R, Turkey) at 25 degrees Celsius for fifteen minutes at three thousand revolutions per minute (3045 xg), the mixture was spun after 24, 48, and 72 hours. After collecting and diluting the liquid that was above the sediment, it was then subjected to HPLC analysis [17].
HPLC Analysis
Additionally, high-performance liquid chromatography (HPLC) and other literature data were used in order to create and validate the VST quantification approach in the SEDDS and S-SEDDS formulations. This was accomplished with the assistance of an Agilent (HP 1100, USA) Series of equipment. Using a Zorbax SB C18 column that measured 150 mm × 4.6 mm and had a particle size of 3.5 µm, an experiment set up at 25 °C was carried out. The mobile phase was prepared by adding trifluoroacetic acid at a flow rate of 1.0 mL/min. The volume-to-volume ratio of acetonitrile to 0.1M phosphate buffer was also adjusted. The pH was then reduced to 2.7. An ultraviolet-visible detector was used to assess the absorbance at a 250 nm wavelength in order to identify VST [18,19].
SEDDS Preparation
In order to find out whether the SEDDS formulation is the best, a pseudo-ternary phase diagram is necessary [20]. The oil, surfactant, and co-surfactant were mixed using the water titration method to get the most effective formulations. These formulae generated the most area due to their application, as seen by the program's center of gravity. The microemulsion area was determined using phase diagrams created using [21], and the oil, surfactant, and cosurfactant combinations were selected after the SEDDS region was identified by water titration. The oil, surfactant, and co-surfactant were combined in a magnetic stirrer set to fifty revolutions per minute (rpm). The mixture was then heated to thirty-seven degrees Celsius (°C) to create a transparent solution. The oily mixture was then supplemented with half a milliliter of VST, which contained 80 milligrams in total. Dissolving the VST in a transparent solution required 50 revolutions per minute of stirring with a magnetic stirrer. The answer was produced by doing this.
Characterisation of VST-SEDDS
This research looked at a wide range of physicochemical aspects of the VST-SEDDS mixture. From the outside, people looked in. The pH meter, conductometer, and refractometer used to measure the VSTSEDDS formulation were all instruments manufactured by Mettler Toledo in Switzerland, Jenway in England, and Kruss in Germany, respectively. A Brookfield DVII + Pro viscosimeter (made in the US) was used to measure the formulation's viscosity at a temperature of 37 ± 1 ºC. The VST-SEDDS formulation's droplet size and zeta potential values were determined using a Malvern Nano ZS instrument operating at a temperature of 25 ± 2 ºC. Furthermore, the appropriate amount of water was determined by consulting the ternary phase diagram. The SEDDS VST content was quantified using a conventional method using HPLC. A temperature of 37±0.5°C was used to add formulations dropwise to 1000 mL of distilled water for the purpose of visually assessing the emulsification time. Utilizing a dissolving apparatus classified as USP (XXII) class II, the mixture was vigorously mixed at a rate of 100 revolutions per minute. Centrifugation, a freezing-thawing cycle, and a heating-cooling cycle were used to investigate the thermodynamic stability of VST-SEDDS [22].
S-SEDDS Preparation
In order to adsorb SEDDS formulations that included VST onto inert carriers (namely Avicel pH101 and HPMC, respectively), wet granulation technology was used. The dry mixture was produced by placing it in an oven that was preheated to 45 degrees Celsius for about one hour. By loading varying amounts of formulation into inert carriers, we were able to determine the compositions of the optimum formulations.
Characterisation of VST-S-SEDDS
There were other factors that were taken into consideration, including dimensional analysis using a vibrating screen (Retsch, Germany), emulsification time, particle size, bulk density, and tapped density (Logan Tap 2S, India).
Stability Chemically and Physically
The stability of the S-SEDDS formulation was tested using two different formulations. The formulations were maintained for a period of twelve months at two different temperatures and relative humidity levels: 25±2˚C and 60±5%, respectively. To conduct the research on stability, the samples were observed for a period of twelve months beginning at time t=0, which was the beginning of the study, and then again at time intervals of one, three, six, nine, and twelve months. In order to determine whether or not S-SEDDS formulations that include VST can be successfully administered in hard gelatin capsules (00), a stability study was carried out.
In Vitro Release Studies
All of the VST-SEDDS, VST-S-SEDDS, commercial formulation, and powder VST were subjected to in vitro release experiments at 37 degrees Celsius (with a standard variation of 1 degree Celsius) and 50 rotations per minute. The trials were conducted using the spinning paddle method. pH values of 1.2, 4.6, and 6.8 were utilized in combination with FaSSIF, FeSSIF, and FaSSGF, respectively, on the scale. Each formulation underwent three independent parallel release experiments throughout the research. After the mixtures were mixed with 400 milligrams of VST per milliliter, they were sealed in firm gelatin capsules labeled with the number 00. At 0, 5, 10, 20, 30, 45, 60, 90, 120, 180, 240, 300, 360, 420, 480, and 1440 minutes, samples were obtained from the dissolving medium. Each of these periods was used to acquire the samples. After the supernatant was filtered to remove particles larger than 0.45 μm, its concentration was measured using the high-performance liquid chromatography (HPLC) method [23].
Statistical Evaluation
We performed statistical analysis on the data that we gathered in the laboratory. The data were examined according to whether they were comparable or different from one another. A Student t-test or an analysis of variance (ANOVA) was the most effective method for analyzing parametric tests, and a Kruskal-Wallis test was the most effective method for analyzing nonparametric tests. With a p-value below 0.05, statistical significance was established.
Result And Discussion
Screening Tests
Researchers employed a variety of oils, surfactants, and co-surfactants in their examination of solubility. Based on the data provided, we have determined that our formulation studies must investigate the best dosages of the following ingredients: isopropyl myristate (oil phase) at 3.5 mg/mL, the sufactans Capyrol 90 (19.8 mg/mL) and Tween 20 (32.5 mg/mL), and the co-surfactant Transcutol HP (168.9 mg/mL) (Table 1).
Table 1. Solubility Tests of VST in Three Different Phases: Oil, Surfactant, and Water
|
Phase / Excipient |
Solubility of VST (mg/mL) ± SD |
|
Oils |
|
|
Oleic Acid |
3.20 ± 0.023 |
|
Soybean Oil |
2.40 ± 0.010 |
|
Isopropyl Myristate |
3.50 ± 0.570 |
|
Surfactants |
|
|
Capryol 90 |
19.79 ± 0.890 |
|
Labrafil M 1944 CS |
17.50 ± 0.600 |
|
Span 80 |
9.80 ± 1.100 |
|
Tween 20 |
32.45 ± 0.870 |
|
Tween 80 |
24.50 ± 3.400 |
|
Cremophor EL |
32.34 ± 1.980 |
|
Co-surfactants |
|
|
PEG 600 |
93.43 ± 5.200 |
|
Transcutol HP |
168.90 ± 6.400 |
|
Aqueous Phases |
|
|
Water |
0.09 ± 0.001 |
|
pH 1.2 buffer |
0.04 ± 0.004 |
|
pH 6.8 buffer |
3.87 ± 0.056 |
HPLC Analysis
A method based on high-performance liquid chromatography (HPLC) was developed and shown to be useful for determining VST release studies in vitro. In order to compare the validation method's findings with those of the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH), we performed an analysis. The parallels and dissimilarities between the two data sets were the primary foci of this study. An in vitro release-optimal linearity method is warranted, as shown by the three analytical curves. This technique is applicable throughout a concentration range of 0.5-50 µg/mL. We employed the linear regression equation that we acquired using the least squares method in order to calculate the correlation coefficients; the results are shown in Table 2. An average recovery of between 98.00% and 102.00% was observed at concentrations of 80, 100, and 120 µg ml−1. Every single one of the relative standard deviations, the value of the average precisions, and the accuracy was all lower than 2%. All of the extra validation criteria were found to be within acceptable ranges on their own.
Table 2. LOD, LOQ, and Linearity
|
Medium |
Concentration Range (µg/mL) |
Equation |
R² |
LOD (µg/mL) |
LOQ (µg/mL) |
|
pH 1.2 |
0.5 – 50 |
y = 17.400x + 5.3110 |
0.9999 |
0.070 |
0.210 |
|
pH 4.5 |
0.5 – 50 |
y = 18.015x + 2.1077 |
0.9991 |
0.027 |
0.081 |
|
pH 6.8 |
0.5 – 50 |
y = 17.692x + 0.6017 |
0.9999 |
0.031 |
0.093 |
|
FaSSIF |
0.5 – 50 |
y = 17.606x – 1.2497 |
0.9999 |
0.035 |
0.105 |
|
FeSSIF |
0.5 – 50 |
y = 17.813x – 7.6681 |
0.9994 |
0.029 |
0.087 |
|
FaSSGF |
0.5 – 50 |
y = 16.815x – 6.3273 |
0.9991 |
0.060 |
0.180 |
VST-SEDDS Preparation and Characterization
When it comes to o/w emulsions, it is usual practice to need an HLB value between 10 and 14, which indicates a stronger hydrophilic lipophilic balance [24]. When developing several SEDDS formulations, different surfactant/co-surfactant ratios were used (1:0.5, 1:1, and 1:1.5). The HLB value of the o/w emulsion was taken into account throughout the development process. With regard to the extensive microemulsion zone, the ratio of surfactant to cosurfactant was set at 1:1, as seen in Figure 1.
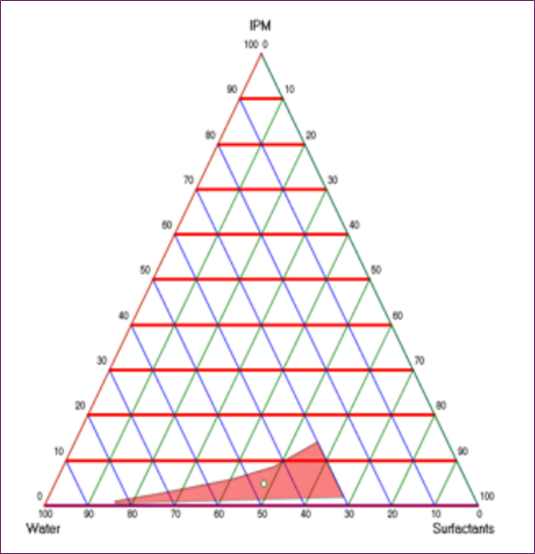
Figure 1. SEDDS Pseudo-Ternary Phase Schematic With 1:1 Surfactant/Co-Surfactant Ratio
An 80 mg/0.5 mL solution of VST was prepared in SEDDS. For see-through VSTSEDDS, a phase shift was unnecessary. Physical analysis revealed that the VSTSEDDS formulation was devoid of phases, homogeneous, and cloudiness. The pH of VST-SEDDS was 3.74. The electrical conductivity of the formulation was 89 µS/cm. The emulsion is o/w according to these numbers [25]. AT 150 rpm, VSTSEDDS exhibits a viscosity of 47 cP and a torque of 98%. The refractive index was 1.45 while the size of the droplets was being studied. With a zeta potential of 0.069 ± 0.005, the average size of the droplets was 105.2 ± 15 nm. The dispersion of particle sizes is quantified by the polydispersity index (PDI). A previous research found that a PDI less than 0.05 suggests a homogenous system with a restricted dispersion of particle sizes [26, 27]. A PDI of 0.2 is seen in the homogenous VST-SEDDS formulation. The quantity was measured at 100.2% using an approved HPLC technique. The effectiveness of the system is shown by the emulsification time in self-emulsification [28]. Balakumar et al. [29] discovered that formulations were stable after emulsification took less than one minute. Within 10 seconds, VST-SEDDS was fully disseminated, according to the literature. A study was conducted to determine the formulation's thermodynamic stability [30]. Stabilization with VST-SEDDS was unaffected by phase separation or precipitation.
S-SEDDS Preparation and Characterization
Wet granulation was used to successfully separate and convert VST-containing SEDDS formulations into Avicel pH101 and HPMC. We were able to effectively obtain white, homogeneous pellets that looked like S-SEDD. With bulk density (weight/initial volume) and tapped density (weight/tapped volume) as indicators of packing ability, the Carr index (%difference of final and initial density relative to final) and the Hausner ratio (final/initial density) were computed, as shown in Table 3. Both of these proportions derive from the end density to beginning density ratio. These findings indicate that the granule flow was inadequate. The VSTSEDDS-HPMC approach obtained a success rate of 94% in dimensional analysis, whereas the VST-SEDDS-Avicel technique generated a success rate of 98.4% within the 2000-1400 µm mesh size range. These findings pave the way for the production of homogenous grains. Unlike the HPMC, which took five minutes to administer, VST-SEDDS-Avicel only took thirty seconds. According to the measurements, the particles of VST-SEDDS-Avicel were 186.3±1.362 nm in size, whereas those of VST-SEDDS-HPMC were 129.1±5.36 nm. Granulation caused a little rise in droplet size.
Table 3. “Flow Properties of Solid S-SEDDS
|
Formulation |
Bulk Density (g/mL) |
Tapped Density (g/mL) |
Hausner Ratio |
Carr’s Index (%) |
|
VST-SEDDS-Avicel |
0.288 |
0.4301 |
1.492 |
32.98 |
|
VST-SEDDS-HPMC |
0.222 |
0.3080 |
1.389 |
27.9 |
Stability Chemically and Physically
Over the duration of precisely one year, the VST-SEDDS-Avicel and VST-SEDDS-HPMC formulations underwent a battery of chemical and physical tests. For these investigations, the temperature and relative humidity were set at 25±2 degrees Celsius and 60±5%, respectively. In contrast, the other two studies employed 40±2 degrees Celsius and 75±5%, respectively. After one year, the active ingredient content and physicochemical tests were revisited, and the findings revealed that neither of the stability settings had changed significantly.
In vitro Release Studies
In vitro release studies are conducted in a controlled environment to assess the effectiveness of newly developed formulations [31]. These studies are very important for predicting the release of drugs in vivo. We were able to do this by testing the powdered VST, commercial formulation, VST-SEDDS, and VST-S-SEDDS with in vitro release at pH1.2,4.6, and 6.8. The amount of the chemical released was determined by these parameters. Because of its acidic nature, VST has a bioavailability of twenty-five percent. The substance is not highly soluble when the pH is low, and it may be absorbed in the upper gastrointestinal system at a very low level [32]. As a result, it is of the utmost importance to make certain that the drug delivery system that has been developed improves its solubility at a low pH (pH 1.2). It was found that the commercial formulation released 62.9 percent, and the VST powder released 9.4 percent after 24 hours of in vitro release experiments at a pH of 1.2. On the other hand, the VST-SEDDS formulation released 90.7 percent, the VSTSEDDS-Avicel formulation released 82.4 percent, and the VST-SEDDS-HPMC formulation released 77.2 percent (Fig. 2). It has been shown via this discovery that the SEDDS and S-SEDDS formulations enhanced the solubility of VST at a pH of 1.2, which resulted in an increase in the amount of VST that was released. The results of in vitro release studies that were carried out at pH 4.6 and pH 6.8 correspond to the figures shown in Figure 2-B and 2-C, respectively. Formulations including VSTSEDDS-HPMC showed an increase in solubility at pH 4.6 and 6.8, in contrast to powder VST. When compared to powder VST, this was seen. Its solubility was equivalent to that of the commercial product. When compared to VST-SEDDS and VST-SEDDS-Avicel, the commercial formulation exhibited a lower in vitro release in a variety of media. Furthermore, the release profile of VST-SEDDSHPMC, which is an S-SEDDS formulation, was slower than that of VST-SEDDS-Avicel across all pH levels. [33] This is because HPMC decreased the quantity of medicine that was released from the matrices, which is the reason for this result.
The physiological ambient pH values that are provided by guidelines for low solubility active chemicals do not accurately reflect the conditions that exist in their natural environment. As a consequence of this, biocompatible environments that are intended to provide conditions that are comparable to those found in the gastrointestinal system have been developed. It is dependent on whether or not the person is fasting that the composition, osmolality, pH, and buffer capacity of the medium are affected [34]. Figure 2-D, 2-E, and 2-F show the results of the dissolving tests that were conducted under fed and fasting conditions using VST powder, commercial formulation, VST-SEDDS, and VST-SEDDSAvicel, respectively. No studies have been conducted on the topic due to the delayed-release properties of HPMC. A fed state rise of 44.57%, 79.82%, and 99.55% respectively, enhances the release in the produced VST-SEDDS formulation. The digestive system contains several amphiphilic molecules, which greatly affect the feeding condition, solubility, dissolving rate, and absorption of some medications [35]. This is why this happens. Chemicals such as bile salts, phospholipids, monoglycerides, and fatty acids are all part of this class.
Contrarily, the obtained VST-SEDDS-Avicel remains constant whether the patient is either fed or fasted (FaSSGF 37.57%, FaSSIF 100.16%, and FeSSIF 98.39%). Therefore, except for the specifically targeted acidic medium, the solubility of the SEDDS formulations was seen to have improved in all dissolving media. It was only the VST-SEDDS-HPMC formulation that changed the release profile, which led to a slower release rate. This occurred because the polymer structure of the formulation was different from the other formulations. The release patterns of VST-SEEDS and VSTSEDDS-Avicel were statistically equivalent (p>0.05) at pH1.2, pH4.6, pH6.8, FaSSGF, and FeSSIF. This was the case at many different pH levels. VST-SEDDS-Avicel exhibited superior performance in terms of release rate when compared to VST-SEDDS in FASSIF medium. This was due to the fact that its solubility was not influenced by pH (p<0.05).
In a previous study that investigated both pharmacokinetics and pharmacodynamics, the bioavailability of the SEDDS formulation was shown to be 423% higher than that of the commercial version. On the basis of the release properties and pH-independent solubility of the S-SEDD formulation, this study anticipated a PK response that is either equivalent or better. We want to undertake in vivo testing utilizing a tablet dosage formulation that has excipients added to it in order to make it more compressible. This is something that we want to accomplish.
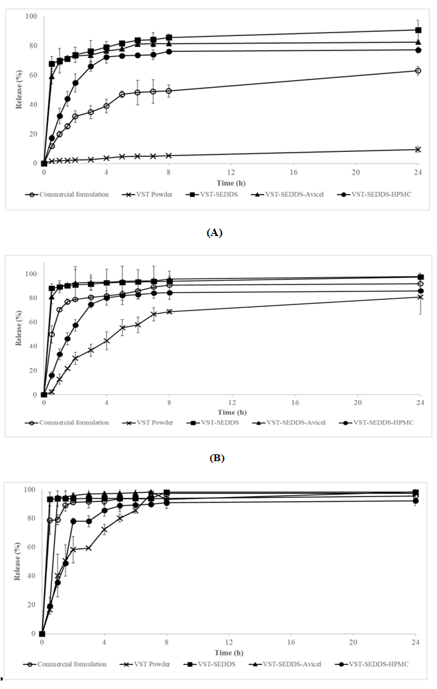
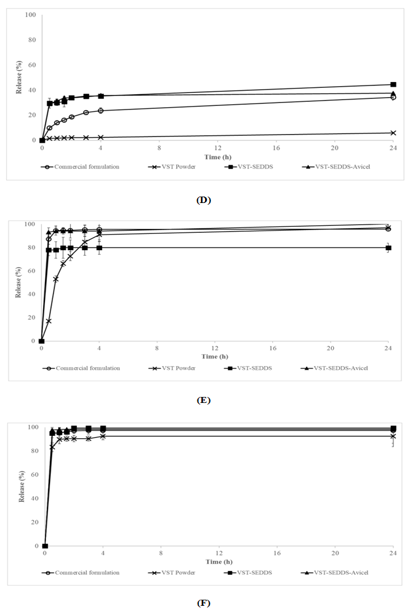
Figure 2. “Release Profiles of VST Powder, SEDDS, and Commercial Formulations in Different Media (FaSSGF, FaSSIF, FeSSIF, pH 1.2, 4.6, 6.8, 7.8)
Conclusion
When administered in different media, VST powder, SEDDS, and commercial formulations exhibit the following release profiles: pH1.2,4.6,6.8, and 7.8; FaSSGF, FaSSIF, and FeSSIF. During the course of this work, valsartan SEDDS formulations in both liquid and solid forms (S-SEDDS) were successfully produced and described. In comparison to the product that is now available on the market, the changed formulations significantly improved drug release in acidic conditions. This was made possible by the low pH solubility of valsartan. It was the combination of VST, SEDDS, and Avicel that demonstrated the most favorable release properties, including stability both when the patient was fed and when they were fasting, as well as a solubility that was not affected by pH. In light of these findings, it is fair to infer that S-SEDDS and valsartan-loaded SEDDS might be viable alternative dosage forms for improving hypertension therapy. This would be accomplished by reducing the limits that the standard formulation imposes on solubility and bioavailability
- World Health Organization. (2021). Hypertension fact sheet. WHO.
- Whelton,P.K.(2018).2017ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults. Journal of the American College of Cardiology. 71(19): 127–248.
- Kearney, P. M. (2005). Global burden of hypertension: analysis of worldwide data. The Lancet. 365(9455): 217–223.
- Amidon, G. L. (1995). A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharmaceutical Research. 12(3): 413–420.
- Dahan, A.Miller, J. M. (2012). The solubility–permeability interplay and its implications in formulation design and development for poorly soluble drugs. AAPS Journal. 14(2): 244–251.
- Lipinski, C. A. (2002). Poor aqueous solubility—an industry wide problem in drug discovery. American Pharmaceutical Review. 5(3): 82–85.
- Burnier, M, Brunner, H. R. (2000). Angiotensin II receptor antagonists. The Lancet. 355(9204): 637–645.
- Israili, Z. H. (2000). Clinical pharmacokinetics of angiotensin II (AT1) receptor blockers in hypertension. Journal of Human Hypertension. 14(1): 73–86.
- Al Omari, M. M. H. (2001). Dissolution, stability and solubility of valsartan in different aqueous media. Journal of Pharmaceutical and Biomedical Analysis. 25(3-4): 487–492.
- Goyal, R. (2011). Solubility enhancement of valsartan using solid dispersion. International Journal of Pharmacy and Pharmaceutical Sciences. 3(1): 72–76.
- Pouton, C. W. (2006). Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. European Journal of Pharmaceutical Sciences. 29(3–4): 278–287.
- Tang, B. (2008). Development of solid self-emulsifying drug delivery systems: Preparation techniques and dosage forms. Drug Discovery Today. 13(13–14): 606–612.
- Porter, C. J. H. (2008). Lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nature Reviews Drug Discovery. 7(3), 231–248.
- Singh, B., et al. (2012). Solid self-emulsifying drug delivery systems (S-SEDDS): An overview. Asian Journal of Pharmaceutics. 6(4): 209–217.
- Balakrishnan, P. (2009). Solid self-emulsifying drug delivery system (S-SEDDS) for improved oral delivery of poorly soluble drugs. Bulletin of the Korean Chemical Society. 30(2): 329–336.
- Singh, A. K. (2018). Self-emulsifying drug delivery systems for improved oral bioavailability of lipophilic drugs: A review. International Journal of Pharmacy and Pharmaceutical Sciences. 10(8):1–7.
- Diril, M, Türkyılmaz, G. Y, Karasulu, H. Y. (2019). Formulation and In Vitro Evaluation of Self Microemulsifying Drug Delivery System Containing Atorvastatin Calcium. Current Drug Delivery. 16(8): 768–779.
- Shirkhedkar, A. A, Chaudhari, S. R, Patil, A. S, Surana, S. J. (2017). A concise review on analytical profile of valsartan. Eurasian Journal of Analytical Chemistry.12(4): 337–364.
- Zarghi, A, Shafaati, A, Foroutan, S. M, Movahed, H. (2008). Rapid quantification of valsartan in human plasma by liquid chromatography using a monolithic column and a fluorescence detection: Application for pharmacokinetic studies. Scientia Pharmaceutica. 76(3): 439–450.
- Khan, F, Islam, M. S, Roni, M. A, Jalil, R. U. (2012). Systematic development of self-emulsifying drug delivery systems of atorvastatin with improved bioavailability potential. Scientia Pharmaceutica. 80(4): 1027–1043.
- Gülmezoğlu, E, Yıldız Türkyılmaz, G, Karasulu, H. Y. (2022). Preparation and evaluation of a lipid-based drug delivery system to ımprove valsartan oral bioavailability: pharmacokinetic and pharmacodynamic analysis. Drug Development and Industrial Pharmacy. 48(12): 727–736.
- Jianxian, C, Saleem, K, Ijaz, M, Ur-rehman, M. (2020). Development and in vitro Evaluation of Self-emulsifying Drug Delivery System. International journal of Nanomedicine. 15: 5217–5226.
- Tadikonda, R. R, Ade, S. (2020). Preparation and in vitro evaluation of sustained release bilayered matrix tablets of valsartan. World Journal of Pharmacy and Pharmaceutical Sciences. 9(6): 1307–1319.
- Park, Y. H, Kim, H. J. (2021). Formulation and stability of horse oil-in-water emulsion by HLB system. Food Science and Biotechnology. 30(7): 931–938.
- Thakkar, H, Parmar, M, Nangesh, J, Patel, D. (2011). Formulation and characterization of lipid-based drug delivery system of raloxifene-microemulsion and self-microemulsifying drug delivery system. Journal of Pharmacy and Bioallied Sciences. 3(3): 442.
- Alwadei, M, Kazi, M, Alanazi, F. K. (2019). Novel oral dosage regimen based on self-nanoemulsifying drug delivery systems for codelivery of phytochemicals – Curcumin and thymoquinone. Saudi Pharmaceutical Journal. 27(6): 866–876.
- Tzeyung, A. S, Md, S, Bhattamisra, S. K, Madheswaran, T, Alhakamy, N. A, Aldawsari, H. M, Radhakrishnan, A. K. (2019). Fabrication, optimization, and evaluation of rotigotine-loaded chitosan nanoparticles for nose-to-brain delivery. Pharmaceutics. 11(1): 1–17.
- https://wjpr.s3.ap-south-1.amazonaws.com/article_issue/1404208762.pdf
- Balakumar, K, Vijaya, C, Tamil, N, Hari, R, Abdu, S. (2013). Colloids and Surfaces B : Biointerfaces Self nanoemulsifying drug delivery system (SNEDDS) of Rosuvastatin calcium: Design, formulation, bioavailability and pharmacokinetic evaluation. Colloids and Surfaces B: Biointerfaces, 112(2013): 337–343.
- Panner, S. R, Kulkarni, P. K, Dixit, M. (2013). Preparation and evaluation of selfnanoemulsifying formulation of efavirenz. Indian Journal of Pharmaceutical Education and Research. 47(1): 47–54.
- Varela-Moreira, A, van Leur, H, Krijgsman, D, Ecker, V, Braun, M, Buchner, M, Fens, M. H. A. M. (2022). Utilizing in vitro drug release assays to predict in vivo drug retention in micelles. International Journal of Pharmaceutics, 618(December 2021): 121638.
- Aydın, Z, Öztürk, S. (2014). Hipertansiyon tedavisinde güncel yaklaşımlar. Haseki Tip Bulteni. 52(4): 251–255.
- Alǧin, E, Kiliçarslan, M, Karataş, A, Yüksel, N, Baykara, T. (2004). Effects of polymer type, polymer:direct tabletting agent ratio and tabletting method on verapamil hydrochloride extended release from hydroxypropylmethylcellulose matrix tablets. Ankara Universitesi Eczacilik Fakultesi Dergisi. 33(3): 125–137.
- Sarisaltik Yaşin, D, Teksı̇n, Z. Ş. (2018). Evaluation of biorelevant dissolution media simulating fasted and fed states. Fabad Journal of Pharmaceutical Sciences. 43(3): 217–232.
- Kleberg, K, Jacobsen, J, Müllertz, A. (2010). Characterising the behaviour of poorly water soluble drugs in the intestine: Application of biorelevant media for solubility, dissolution and transport studies. Journal of Pharmacy and Pharmacology. 62(11): 1656–1668.
Download Provisional PDF Here
PDF