

p (1).png)



.png)




.png)

Review Article
The Language of Proteins, And Protein Structure
- Bruno Riccardi
Corresponding author: Bruno Riccardi Graduated in Biology, La Sapienza University of Rome.
Volume: 2
Issue: 4
Article Information
Article Type : Review Article
Citation : Bruno Riccardi, The Language of Proteins, And Protein Structure. Journal of Medical and Clinical Case Reports 2(4). https://doi.org/10.61615/JMCCR/2025/AUG027140822
Copyright: © 2025 Bruno Riccardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI: https://doi.org/10.61615/JMCCR/2025/AUG027140822
Publication History
Received Date
02 Aug ,2025
Accepted Date
15 Aug ,2025
Published Date
22 Aug ,2025
Abstract
This article is a review of the fundamental properties of proteins.
As such, it describes and examines proteins in all their structural and functional aspects, which are already known and widely understood, but it proposes an original interpretation in identifying the endogenous and exogenous causes of their transformations that determine the incorrect folding, the misfolding, which is at the basis of their pathogenic behavior.
From prion diseases to amyloid diseases, we want to identify the trigger factors that determine protein misfolding and the consequent pathogenic effect.
The article also aims to investigate the intrinsic cause that determines the breakdown of the defective proteins into their healthy counterparts and their accumulation within the parenchyma. This accumulation is the recognized efficient cause of many diseases.
The article is a concise review of already known knowledge that adds only a few considerations on the fundamental role of protein structures.
Keywords: Amino acids, Protein structures, Misfolding, Prions, Protein synthesis, Intrinsically disordered proteins, Factors that alter protein structures.
►The Language of Proteins, And Protein Structure
Bruno Riccardi1*
1Graduated in Biology, La Sapienza University of Rome.
Introduction
In this article, we use a particular key to understand the meaning of protein structures.
The informational power of proteins, which is their most salient characteristic, depends on the spatial and temporal arrangement of their individual constituent elements.
Individual proteins are the result of millennia of selection that have led each of them to have a biological significance that is activated when they come into steric contact with other structures and modify their behavior.
Their great versatility lies in the fact that they can take on the most varied forms and, as their size increases, their information content enriches.
For these reasons, they cannot be replaced by equivalent structures.
Proteins are a class of compounds of primary importance for living organisms.
They play a central role in all the structural and functional biological aspects of the plant and animal kingdom, so much so that a minimal variation in their natural composition and structural conformation leads to dysfunction and cell death [1-2].
No other molecule has the same capacity to achieve conformational complexity with the possibility of equal informational content as proteins.
Proteins can assume four different levels of structural/conformational organization [3-4]:
Primary, secondary, tertiary, and quaternary structures Fig.4 represent their arrangement in the steric and energetic information that they assume in space.
A single amino acid addition or substitution of the primary structure is sufficient to determine a change in the derived structures and can lead to new evolutionary changes.
To date, 42 million proteins have been sequenced, and their amino acid composition and crystalline structure are known
(AlphaFold: https://alphafold.ebi.ac.uk/ ).
General Properties of Proteins
Amino Acids
To understand the normal and pathological functioning of proteins, it is necessary to know the characteristics of this fundamental class of compounds.
Proteins are polymers formed from the sequence of 21 basic compounds, which are amino acids.
Amino acids have particular and unique properties, which is why the proteins from which they are made can take on infinite structures and functions.
All amino acids have a basic molecular structure formed by an amino group (NH 2) and a carboxylic group (CO 2) linked to a central carbon to which other molecules of an aliphatic or aromatic nature are linked, which constitute the side chains [5-7], Fig.1.
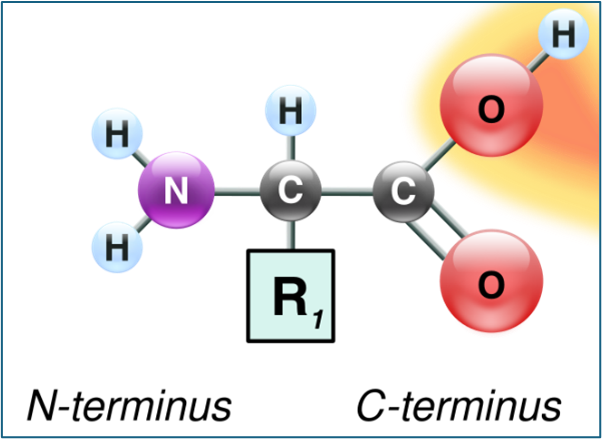
Figure 1 – General Structure of an Amino Acid
Due to their conformation, amino acids have stereoisomeric structures, meaning they are compounds made up of different atoms and molecules bonded to the central carbon, which differ in their spatial arrangement within the molecule. Consequently, the central carbon is an asymmetric chiral carbon, meaning it is bonded to four different substituents.
They all have amphoteric properties, meaning they can bind with acids and bases, and have the nature of zwitterions at physiological pH, meaning they possess both a positive and a negative charge.
This gives amino acids high flexibility and adaptability in the formation of peptide bonds, which can form polymers of unlimited size.
Amino acids can assume the levorotatory (L) and dextrorotatory (D) conformations Fig.2.
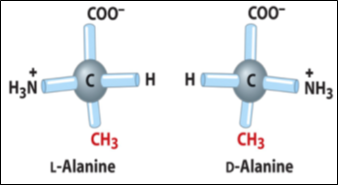
Figure 2- Chiral Structure of an Amino Acid
The 21 amino acids present in proteins are divided based on the functional groups linked to their side chain, which can be:
Non-polar aliphatic (Glycine, Alanine, Proline, Valine, Leucine, Isoleucine, Methionine)
Non-charged polar (Serine, Threonine, Cysteine, Asparagine, Glutamine)
Aromatic (Phenylalanine, Tyrosine, Tryptophan)
Positively charged (basic) (Lysine, Arginine, Histidine)
Negatively charged (acidic) (Aspartate, Glutamate)
The periodic table of amino acids summarizes all the main chemical and structural characteristics of the classes to which they belong. Fig.3.
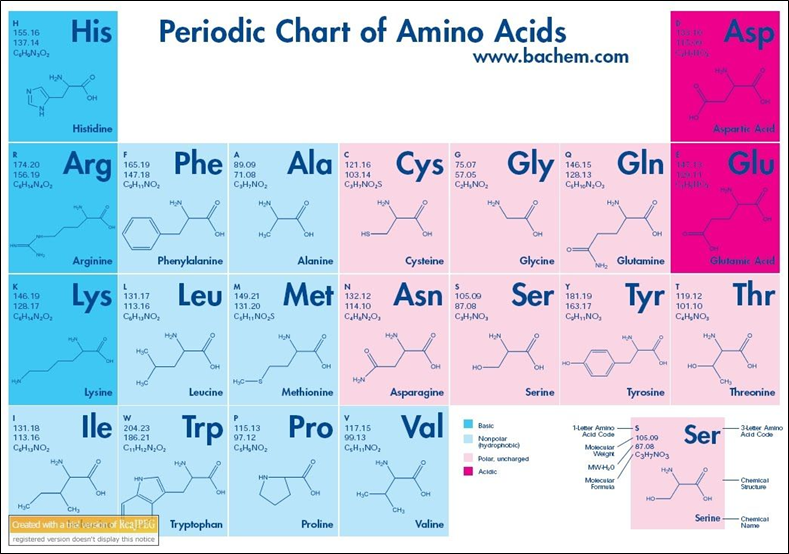
Figure 3- Periodic Chart of Aminoacids
The amino acids present in the periodic table assume a different ionization state based on the pH of the solution in which they are dissolved.
Amino acids are divided into:
Essential nutrients are those that cannot be synthesized by human metabolism and must necessarily be taken in through the diet, and
Non-essential, which can be synthesized through metabolism.
The following amino acids are essential for humans:
Valine, Isoleucine, Leucine, Methionine, Phenylalanine, Tryptophan, Threonine, Lysine
All others are non-essential.
Normally, the amino acids that form proteins have a steric conformation called L, left-handed.
Proteins are formed by the union of amino acids linked together by a peptide bond, a covalent bond, which has the characteristics highlighted in the following diagram:
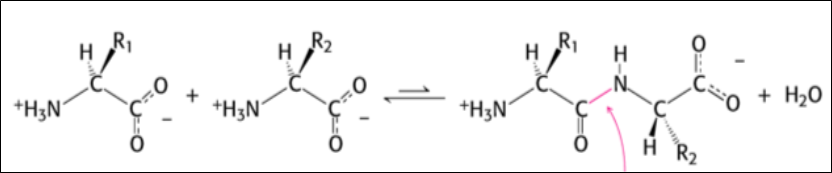
The peptide bond is a polar, planar, and resonance-stabilized bond, and does not allow rotation around the bond, which is why there are Cis and Trans isomeric forms of polypeptides.
Over 99% of peptide bonds in proteins have the TRANS configuration.
Depending on the number of amino acids that form the peptides, we speak of oligopeptides or polypeptides.
Most proteins are polypeptides consisting of 50 to 2000 amino acids.
When proteins are folded into their secondary structure, the amino acids within them form weak bonds, such as hydrogen bonds, electrostatic bonds, and Van der Welt forces. Waals, which stabilize the conformation.
Types of Proteins
Proteins are divided based on their composition into:
Simple proteins: Made up only of a sequence of amino acids;
Conjugated proteins: These are formed by amino acids + metal ions, or non-protein organic molecules, even complex ones. These are glycoproteins (protein + carbohydrates), lipoproteins (proteins + lipids), etc. The non-amino acid part is called the prosthetic group, Table 1.
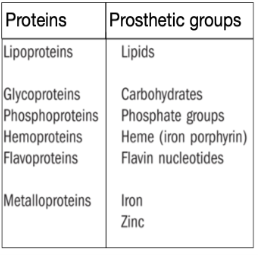
Table 1- Types of Proteins
Based on their Confirmation and the level of structure they assume, proteins are divided into:
Fibrous and Globular
And based on the type of main activity they carry out, they are distinguished into:
Structural Proteins and Functional Proteins, but this distinction is not absolute since most proteins perform both structural and functional functions at the same time.
Furthermore, it must be kept in mind that it is the three-dimensional structure of the protein that has an informational content in itself, and which therefore determines the functional effect.
Protein Structure
As peptide bonds are formed during protein synthesis, proteins tend to spontaneously fold, assuming characteristic spatial arrangements.
For this reason, different structural levels are distinguished: primary structure, secondary structure, tertiary structure, and quaternary structure.
The primary structure is formed by the sequence of amino acids present in the protein. It is of fundamental importance because the protein's function depends on the type and arrangement of amino acids. The primary structure is encoded in DNA.
The secondary structure is represented by the spatial arrangement that the newly formed protein assumes.
Two types of secondary structure can be distinguished: the α-helix form and the β-sheet, which represent the spatial orientation of the protein skeleton, and are determined by the periodicity and the reciprocal spatial arrangement of the amino acids.
Tertiary Structure is produced by the folding of long portions of the protein to minimize steric and energetic hindrance. By virtue of this structure, proteins perform their typical functional role. Folding brings some amino acids distant in the primary sequence closer together. The tertiary conformation can change in response to binding or simple interaction with other molecules.
The quaternary structure, finally, is formed by the union of two or more polypeptide chains that associate through non-covalent interactions.
The quaternary structure is called an oligomer, and the constituent polypeptide chains are called Monomers or Subunits Fig.4.
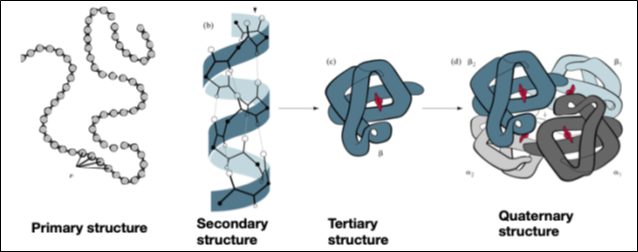
Figure 4- Structure of Proteins
The same protein can be made up of portions arranged in a helix and portions that have a sheet-like shape β. The different portions of the protein play a fundamental biological role in their functioning, and the variation in the type of conformation compared to the natural one leads to the malfunctioning of the protein, with the so-called misfolding, which produces serious pathological effects.
Examples include amyloid formations, present in the brains of patients with Alzheimer's and Parkinson's disease, and prions, etc.
The conjunction between the different portions of the aE lica conformations and Leaflet β is made up of loop segments that do not have a defined structure.
A large group of proteins termed amyloids is the result of misfolding, or incorrect folding, in the secondary structure that involves the transition of the αE portions to the α- helix. in forms Leaflet β. These transformations can affect all organs, with serious pathological consequences.
It is important to emphasize the difference in the amino acid composition of the primary structure of the aE propeller. respect that of Leaflet β.
The most frequent amino acids in α-helices are:
Alanine, Leucine, and Phenylalanine are small or electrically neutral amino acids
Amino acids that are uncommon in α-helices are:
Arginine, glutamic acid, serine, and lysine are polar amino acids that destabilize the helix at physiological pH. Proline is scarce in these forms because it disrupts the helical conformation and prevents its structure from assuming the required shape.
The Alpha-Helix Structure
The aE structure It is the most widespread in proteins.
- It can be right-handed or left-handed, depending on the direction of rotation of the helix, but it is always composed of L-amino acids;
- The left-handed one is less stable due to steric hindrance.
- The right-handed α-helix is the most frequent one in natural proteins Fig. 5.
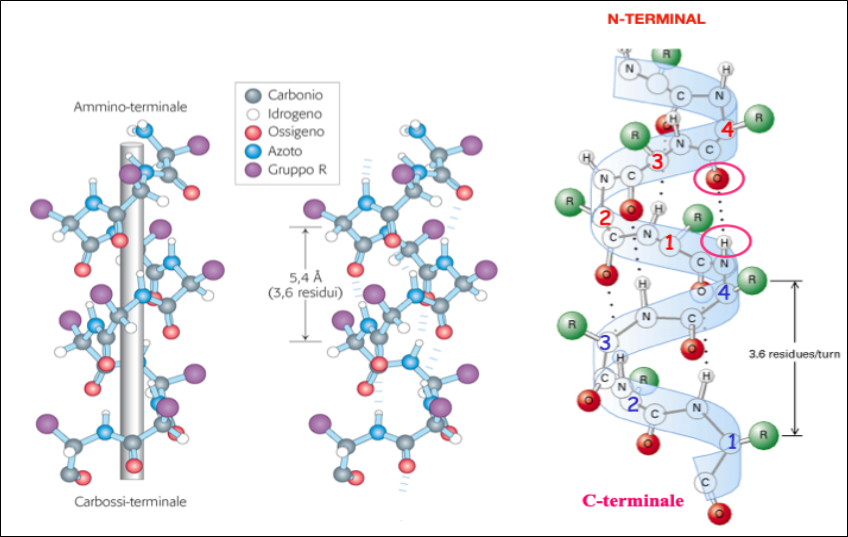
Figure 5- Properties of a-Helical Structures
Leaflet Structure β
Proteins containing these secondary structures exhibit the following properties:
- The polypeptide chains are not coiled in a helix but are almost completely flat - the structure has a folded planar arrangement due to the bond angles.
- Carbonyl oxygen and amide hydrogen protrude almost at right angles to the longitudinal axis of the extended chain.
- Hydrogen bonds are formed between carbonyl oxygen and amide hydrogen of two or more adjacent chains, which stabilize the overall structure.
- The side chains of the various amino acid residues protrude alternately above and below the plane of the folded sheet so that the side chains of adjacent residues are located on opposite sides of the sheet and thus reduce the steric hindrance, Fig. 6.
This secondary structure arrangement is present in several enzymes and in proteins with immune activity.
The pleated β-sheet consists of numerous polypeptide chains arranged adjacent to each other, linked in a continuous structure by short U-shaped sequences.
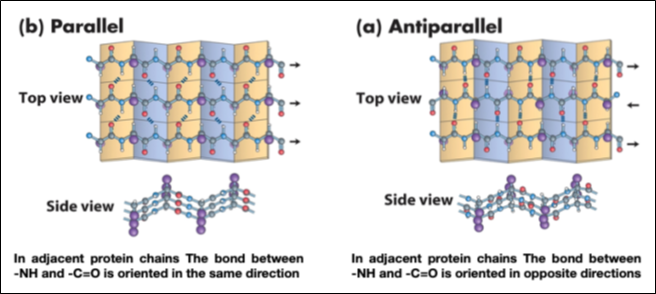
Figure 6- Properties of Sheet β
Some proteins also contain a non-polypeptide part, called a prosthetic group, and are conjugated proteins. Two proteins are called isoforms if, with the same primary structure, they differ in one of the other levels of structure.
Proteins can be denatured by various chemical substances: acids, bases, and by physical means, heat, radiation, or agitation, and assume an unstructured arrangement, which involves the destruction of the spatial conformation and the breaking of hydrogen bonds and disulfide bonds.
A denatured protein, while maintaining its primary structure intact, is no longer able to perform its function unless its tertiary structure is restored, a process called protein renaturation. This property indicates that proteins act through their spatial conformation.
Not all proteins, once denatured, can spontaneously fold back into their original conformation. A protein's conformation, although normally as stable as possible given its amino acid sequence, is not immutable and undergoes small modifications due to interactions with ligands or other proteins. This characteristic underlies the functionality of most proteins.
Many protein complexes that perform important biological functions, such as enzymes, ion pumps, and cellular channels, are formed by protein aggregates joined by non-covalent bonds, which hold the various monomers together while allowing them to assume a different aggregation state in their active form.
The case of hemoglobin is typical.
Protein Folding
All vital functions depend on proteins and, as is known, an alteration in the structure of a protein leads to an alteration or loss of a specific function, as occurs in genetic diseases.
Proteins spontaneously acquire their three-dimensional structure, to which their biological function is inextricably linked.
This process of “structuring” with protein folding (called folding) is encoded in the particular amino acid sequence of each protein.
Continuous monitoring of correct protein folding is performed by protein complexes called "chaperones." There are several classes of chaperones that perform different tasks in correct protein folding.
The fact that chaperones are also of a protein nature raises several questions about the multiple activities of proteins. We are in the presence of complex molecular structures that control the folding of other protein structures, so that proteins can control their own structure. Fig.7.
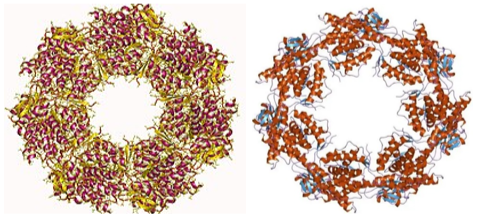
Figure 7 – Different types of Chaperones
How did these proteins acquire this fundamental ability to “direct” the formation of other proteins and regulate their metabolic fate?
Chaperones act in the form of complex systems, but are always formed by protein assemblies, the Proteasome-Ubiquitins.
We will return to this aspect in the section dedicated to the discussion.
Chaperones contribute to the achievement of the 3D structure of functionally active proteins, are able to prevent the formation of a distorted or aggregated structure, and recognize unfolded proteins. Furthermore, they bind to hydrophobic surfaces and inhibit aggregation.
Chaperones are divided into chemical and pharmacological ones.
Chaperones affect the protein folding environment within the cell and stabilize proteins by preventing thermal and chemical denaturation.
chaperones bind to specific protein conformations and stabilize them. They are effective in protecting proteins from proteasomal degradation. Misfolded proteins are eliminated by a complex, ATP-dependent system called the proteasome-ubiquitin system.
The proteasome is a 26S multiprotein complex responsible for degrading polypeptides within the cell. It consists of a core that accepts and then degrades polyubiquitin substrates.
Ubiquitin is a small protein with a regulatory function, present in all cells, hence the name ubiquitin, it determines recognition by the proteasome for degradation.
The general aspects of the mechanism that regulates protein folding are:
- The amino acid sequence, when polypeptide synthesis is completed, the protein folds spontaneously with the assistance of chaperones.
- The cellular environment.
- The correct balance between the various folded parts.
- The efficient functional control of control systems (Chaperones, Proteosome).
Misfolding Protein
The incorrect folding of proteins is called protein misfolding and produces different types of aggregates, which can be distinguished into:
- Proteins that form insoluble aggregates;
They are altered proteins and, for this reason demolished by the proteasome systems, which thus determine a reduction in their number.
A case in point is the misfolding of some functional proteins, which leads to catastrophic consequences due to the metabolic damage produced.
Other examples of polymeric structures besides proteins, such as carbohydrates and lipids, exhibit different behaviors. This indicates that it is not only the structural complexity of molecules that determines their biological characteristics, but also their morphological-conformational adaptation capacity.
In the case of carbohydrates and lipids, the complexity of the polymers is achieved by the sum of similar monomers linked together.
Let's take the case of starch and glycogen. Their degree of structural freedom is limited, just as it is in fatty acids, which have a small margin of upper complexity.
Synthetic polymers also have reduced potential for structural complexity.
A fundamental distinction must be made between the size that the chemical structure of a polymer can assume and its conformational complexity, which can express a high information content.
A few examples will clarify the difference.
For example, sugars. The largest sizes these substances can reach are the polymers starch and glycogen. But even when they reach considerable masses, the degree of information they contain is always limited. Essentially, this property is linked to the fact that the constituent monomers are formed by the same glycidic chemical structure.
The versatility of proteins is different, as it depends on the fact that the possible combinations of their individual components, the amino acids, are extremely high and consist of their sequential arrangement, the primary structure, which can reach an enormous number of possibilities.
Thus, 21 different amino acids can be arranged in 21! (read 21 factorial) sequences with different permutations, where each element can assume different positions in the same molecule.
This explains why proteins are the major component of molecules present in living organisms.
But they have another peculiar characteristic compared to all other classes of molecules: despite the high number of constituent monomers, they organize themselves in space to occupy the minimum volume and the minimum energy content possible.
And this has important implications at the cellular level, where the space available is very limited.
There are some proteins that are secreted outside the cells, in which case they can reach very large sizes, as in silk and lignins.
Speaking of polymers, the case of other complex molecules, such as DNA is noteworthy.
In this case, the constituent elements are only 4, with which 64 possible permutations can be formed, in which the steric arrangement is the same and repeats along the entire length of the molecule.
Since DNA is composed of only four different monomers, which are the nitrogenous bases, its informational content is determined by the sequential arrangement of the constituent monomers, not by their three-dimensional conformation. The structural component that forms the DNA backbone is made up of sugar and phosphate, which are invariably repeated along its entire length.
In other words, the information presented by the various DNA molecules is not direct, as in the case of proteins, but is translated and must be translated into another language with greater informational content, which happens to be in the form of proteins. The arrangement of DNA nucleotides is translated into the primary sequence of proteins.
This means that the informational content of DNA can only be effective if it is present in the structural form of proteins.
The case of RNA is different, as it can also have information content.
Intrinsically Disordered Proteins
There are a particular group of proteins that contain unstable and unstructured amino acid sequences, meaning they assume fluctuating conformations. These sequences, however, can occasionally pair with other polypeptides.
It has been hypothesized that the stable part of the protein may act as a carrier to protect the flexible portion and allow it to pair with other polypeptide sequences, to influence their behavior and modify them [8-10].
It is also hypothesized that these structures could be the origin of prions and amyloid structures and may have played a crucial role in protein evolution.
Disordered proteins have the following properties:
- The unstable portions are contained within other protein structures or are free.
- They do not have a defined structure but are in continuous transformation as if they were trying to reach a definitive conformation.
- Therefore, they must remain in a protected environment, which is constituted by carrier proteins.
Disordered proteins behave like enzymes that transform other proteins with which they come into contact Fig.8.
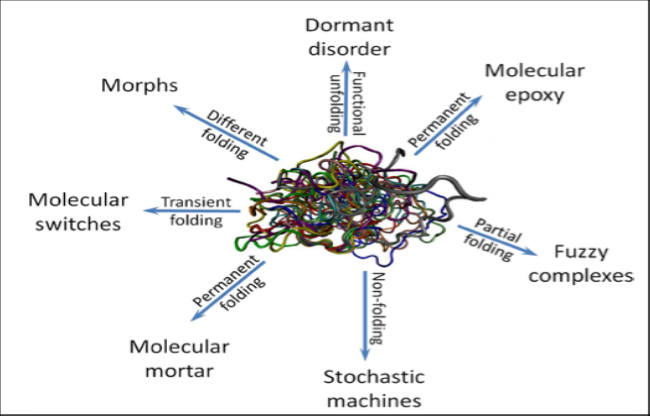
Figure 8 - Schematic Representation of a Variety of Folding Transitions Induced in IDPs/IDPRs by Interaction with Binding Partners. From: Vladimir N. Uversky; Dancing Protein Clouds: The Strange Biology and Chaotic Physics of Intrinsically Disordered Proteins, The Journal of Biological Chemistry Vol. 291, No.13, Pp. 6681–6688, March 25, 2016
The variable component of disordered proteins is made up of amino acids that are continuously shuffled within the carrier protein that contains them, and they continually change their arrangement, collapsing into a stable structure when they encounter complementary polypeptides with which they pair and transform them into other polypeptides or they can mutually exchange amino acids to reach a common equilibrium conformation.
An in-depth comparative analysis of the sequence composition of ordered and disordered proteins revealed that residues such as Ala, Arg, Gly, Gln, Glu, Lys, Pro, and Ser (referred to as disorder-promoting residues) are more frequently present in IDPs (Intrinsically Disordered Proteins) and IDRs (Intrinsically Disordered Proteins). Disordered Regions. Conversely, residues such as Asn, Cys, Ile, Leu, Phe, Val, Trp, and Tyr are more common in ordered/structured segments of proteins (called order-promoting residues).
Comparative studies of amino acid residues in disordered regions, and scales based on physicochemical properties (such as coordination number, aromaticity, propensity to form strands, flexibility index, bulk, propensity to arrange in a helix, etc.) and compositional characteristics have led to the distinction between disordered and ordered regions in proteins.
Misfolding
Misfolding: indicates the incorrect folding of proteins, an event that is at the basis of many human pathologies, collectively defined as misfolding diseases.
The topic of protein misfolding has attracted the attention of many researchers, as it has been found that the phenomenon of incorrect conformational folding of proteins has serious pathological repercussions, due to the irregular functioning of these structures [11-15].
Proteins deformed by misfolding tend to aggregate and form macromolecular complexes that precipitate inside cells and cause cell death or elimination of cells by apoptosis.
The most typical examples of protein misfolding are found in prion forms and amyloid structures, which characterize a complex of pathologies gathered under the common name of amyloid diseases and affect many organs.
The key event that determines the transformation of a normal protein into a misfolded one is represented by the modification of the secondary structure of the aE component, which turns into that of Leaflet β.
The process has been studied in depth by analyzing all the steps that occur during the conversion from a helical secondary structure to a folded sheet structure.
The morphological transformation from native protein to the amyloid form occurs through the passage of an intermediate form of amorphous granular aggregates formed by disordered polypeptide chains.
More than thirty diseases are known that can be traced back to the deposition of these polypeptide aggregates.
They assume particular pathognomonic importance in some pathologies affecting the Central Nervous System, such as Alzheimer's, Parkinson's, Huntington's, and probably also multiple sclerosis etc.
Protein misfolding and aggregation have emerged as a major mechanism in human disease. The list of diseases caused by protein misfolding is growing daily, and as previously mentioned, they are collectively known as amyloid diseases. With aging and other factors, the cell's ability to process and synthesize the proteome, the entire protein structure, declines, becoming a major cause of late-onset diseases (Fig. 9).
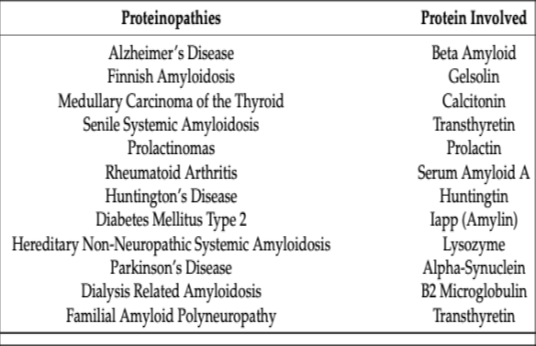
Figure 9- Amyloid-associated proteinopathies. From: Mohammad Rehan Ajmal; Misfolding and Aggregation in Proteinopathies: Causes, Mechanisms, and Cellular Response. Diseases 2023, 11:30
Both nascent and pre-existing proteins are at risk of aggregation in the cytosol. Protein aggregations disrupt cellular function and contribute to aging. Once initiated, the misfolding process reproduces itself exponentially and spreads to neighboring cells.
Deformed proteins are recognized by molecular chaperone systems and eliminated through apoptosis. Mutations and adverse physiological conditions can lead to disorders and pathologies due to incorrect protein aggregation.
chaperones play a central role in maintaining protein homeostasis, guide nascent polypeptides to assume their native structure, and assist them in the translocation and correct folding of denatured intermediates.
Prions
Prions are protein structures observed for the first time in patients who died of brain diseases, who had consumed meat from infected cattle, and were transmitted through a mechanism similar to infectious processes, although they have nothing in common with bacterial or viral forms [16-20].
The pathogenic mechanism of prion infection, also called scrapie infection, was definitively recognized as being caused by protein structures resistant to various therapeutic treatments.
Prion proteins are also present in normal nerve cells and have in their structure the typical a-Elic component. and I am bound to the cell membrane. Their normal physiological function consists of modulating defense against oxidative stress, cell adhesion, and myelin maintenance.
The Cytoskeleton
The cytoskeleton forms the scaffolding of cells and gives them the dynamic morphology that all cells assume during their life cycles. It assists cells in their movement and transfer, as well as providing motor apparatus such as cilia and flagella.
The cytoskeleton includes various protein structures, which are microtubules, microfilaments, and intermediate filaments, which are made up of monomers capable of aggregating and disintegrating into filamentous forms and serve as anchors for cellular organelles in their movements [21-25].
They also intervene in muscle contraction, Fig. 10.
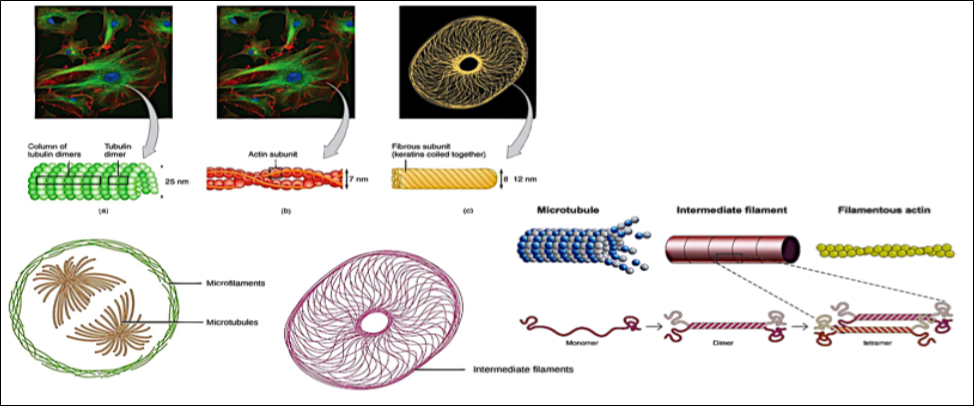
Figure 10 - Structure and composition of the cytoskeleton
This is not the place to describe the individual monomers, a topic that would require several chapters. We mention them only to underline the astonishing properties of proteins.
The cytoskeleton also participates in the formation of the Golgi apparatus with a continuous reciprocal feedback mechanism.
How these proteins manage to assemble the filamentous networks that transport the various organelles and chromosomes in cellular activity and during cell division, and above all, how they manage to follow the correct directions in their movements, remains a mystery.
Here, proteins have reached the maximum level of functional efficiency; not only do they reproduce in individual monomers, but they also know the correct direction to add them to complete the movement. Their ability to adapt and respond to functional demands is astonishing.
They are undoubtedly intelligent molecules.
Since they play a crucial role in the formation of the cytoskeleton, these proteins are also subject to the phenomenon of misfolding, which has direct, disastrous consequences on cellular structure and organization [22-23].
Since they are complex protein apparatuses that derive from the assembly of preformed monomeric proteins, their pathogenic effect has been defined as an aggresome [26-28].
Cell Membrane Proteins
We briefly mentioned the presence of proteins in the cell membrane.
These are proteins bound to the cell membrane that are either entirely contained within the thickness of the lipid bilayer or are attached to other membrane molecules and protrude from the cell surface.
These proteins represent a fundamental component of cells as they are the true effectors of cellular activity, from the transport of substances within the cell, to contact and recognition of other cells or foreign bodies, to receptor function, ion channels, etc.
In fact, the lipid bilayer of the cell membrane serves only to transport proteins to the same level and plays the role of a regulator of overall cellular homeostasis.
Protein Synthesis
To conclude the description of proteins, it's appropriate to mention the mechanism of protein synthesis. A detailed description of the entire process is beyond the scope of this article. We'll simply outline the simplified steps of the entire mechanism.
The mechanism, called "central dogma of Biology" by Watson and Crick, schematically goes through three fundamental stages [29-30]:
- Transcription of the gene that codes for the protein from DNA to messenger RNA, mRNA.
- Transport of amino acids onto mRNA by transfer RNA, Trna.
- Formation of the peptide bond between amino acids.
The process takes place in ribosomes, complex macromolecules present in the cellular endoplasmic reticulum.
Fig. 11 shows a simplified diagram of the process,
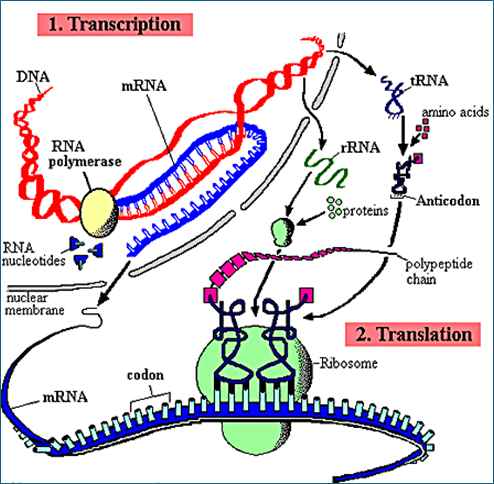
Figure 11 – Schematic Mechanism of Protein Synthesis
Protein synthesis goes through several steps that include the transcription of the protein code from DNA to messenger RNA and the synthesis of the protein at the ribosome level. Fig. 10.
Once synthesized, the native protein undergoes a maturation process that serves to define the definitive sequence that the protein will assume in its target role.
This process is called post-transcriptional control of protein synthesis. During transcription, a nascent protein can be split into two different proteins, a process known as alternative RNA splicing, which can produce different forms of the same protein.
An interesting example of this process occurs in peptide hormones.
Hormones can be derived from lipids (steroid hormones) or from amino acids, as in peptide hormones [31].
Peptide hormones are naturally produced by various endocrine glands in our body. They consist of peptides of varying lengths and are water-soluble; therefore, they can easily travel through the bloodstream to their target cells, where they exert their biological effects.
Hormones are produced in immature forms as preprohormones, which will undergo a maturation process by splicing to become prohormones and then be processed into their final form as hormones [32-33].
This behavior, which recalls that of enzymes, another class of proteins, tells us that the behavior of proteins is very complex and is regulated by multiple biological events that have the task of maintaining the global homeostasis of the entire organism.
How do hormones know the exact sequence they need to interact selectively with their receptors? It's as if they knew in advance the conformation they need to assume to interact precisely with their receptors.
During the protein synthesis process, they know in advance the type and arrangement of amino acids they must contain and how they must be processed after transcription in order to function.
What feedback mechanism regulates the entire process?
These are currently unanswered questions; the only explanation we could venture is that the entire process is the result of the selection of thousands of years of evolution, and once achieved, it has been maintained in all multicellular organisms.
A similar process could have led to the stabilization of metabolic cycles, from glycolysis to the Krebs cycle, which we find identical, with very small variations, in all living things, from bacteria to humans.
Discussion
Proteins have the extraordinary ability to change their conformation in response to effective stimuli to assume a biologically useful transient conformation.
All proteins, from functional to structural (a meaningless distinction), have been selected by biological evolution precisely to fulfill the task of changing their steric conformation based on the needs required by biological activity.
Just to name a few examples:
Enzymes, channel proteins, receptor proteins, cell membrane-bound proteins, the antigen-antibody mechanism, etc.
It is as if they have the ability to assume the required three-dimensional shape when they receive a specific stimulus.
Another aspect that clarifies the function of proteins is the fact that they act by adaptive contact, a sort of formation of a molecular imprint, with the molecules with which they must interact, and only with those.
This means that their conformation is not definitive, and that they are able to modify it.
Therefore, when studying proteins, it is possible to do so through the crystallization process, which causes them to assume a rigid structure, so that they can be studied with X-ray crystallography.
This dynamic behavior can explain the properties of some unusual molecules, such as prions and amyloid structures, which, as we described above, both arise from misfolding compared to what they should have.
This property is probably the key to understanding the behavior of intrinsically disordered proteins that play an important role in the pathogenesis of some diseases.
We can consider these molecules as being made up of a stable polypeptide component in its quaternary structure, which acts as a handle and vehicle that transports them to their final position, while their variable component does not yet have a defined structure.
Then one could think of this variable component as seeking the correct steric coupling with other molecules or metabolites with which it must interact in order to collapse and produce the final functional result.
We can imagine this beady portion, similar to a rubber hose, oscillating and probing the environment in search of other structures with which to mate.
This could be their biological role, and it helps us understand how thermodynamically unstable they are and how susceptible to misfolding and incorrect couplings that produce the most diverse pathologies.
Viewing prions and amyloid structures as intrinsically defective degenerate proteins with the potential to pair with and deform other similar proteins would provide us with clues for attempting to halt the degenerative process, and perhaps even the diseases they cause.
Let us ask ourselves then: where does the ability of misfolded proteins to modify their structure and promote the deformation of other similar proteins in the modified form come from?
Misfolded proteins that we know best is the transition of the alpha-helical portions into beta- sheet forms (Fig. 12). When they assume this shape, they are no longer recognized by the chaperone system and the immune system and are free to act undisturbed, allowing them to assume conformations capable of sterically coping with similar molecules and producing their conformational distortion. Essentially, they function as enzymes.
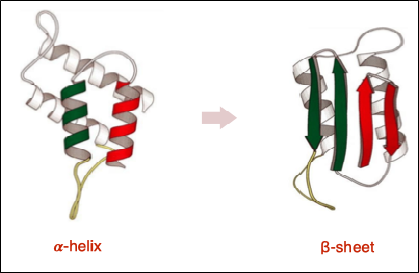
Figure 12 - The conformational transition from alpha helix to beta sheet exposes hydrophobic amino acid residues and promotes protein aggregation
Factors that Alter Protein Structures
In addition to the events described above that intervene in the conformational transformation of proteins from native to their misfolded forms, there are other causes capable of determining their deformation. In particular, some substances have clinical and pathological relevance in the pathogenesis linked to their ability to induce protein misfolding.
Some of these substances have assumed the value of pathogenic markers in clinical diagnosis.
Here we highlight just three of these substances: homocysteine, glucose, and cholesterol. These substances, when present in excess in the blood, are considered important risk factors.
Homocysteine (Hcy) and N- Homocysteinylated Proteins
Homocysteine is a sulfur-containing amino acid, chemically it is a γ– thio –α– aminobutyric acid (C 4 H 9 NO 2 S), which is formed, as an intermediate product, in the enzymatic transformation of methionine (C 5 H 11 NO 2 S) into cysteine (C 3 H 7 NO 2 S), through the removal of methyl groups: the prefix homo- (derived from the Greek ὁμο -) indicates similarity between two compounds [34].
The conversion of homocysteine to methionine (a process called remethylation) or its conversion to cysteine (transsulfuration) represents the main metabolic pathways capable of maintaining its intracellular levels within a narrow range; its release into the bloodstream allows us to measure its plasma concentrations, which represent an index of the tissue homocysteine status Fig.13.
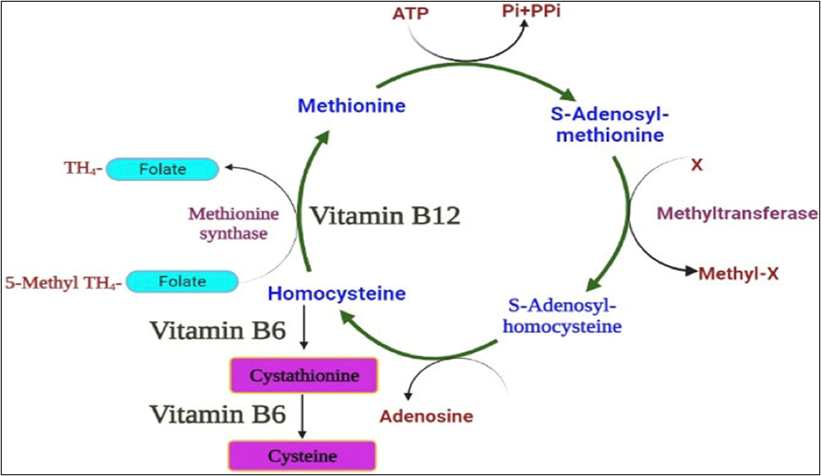
Figure 13 – Homocysteine Metabolism
Homocysteine levels in the blood must be kept under strict control in order to prevent the numerous pathogenic effects reported in the literature and schematically indicated in Fig.14.
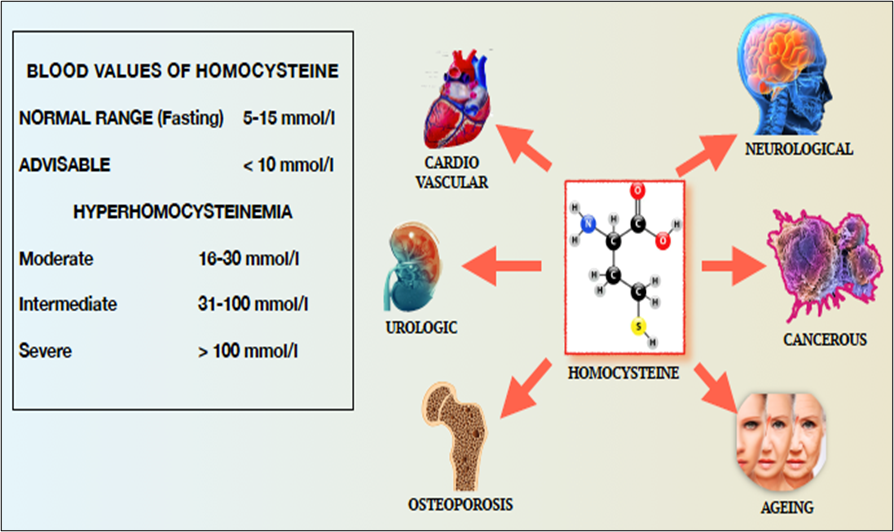
Figure 14 – Blood Values of Homocysteine and Pathologies Resulting from Hyperhomocysteinemia
Homocysteine, present in high concentrations in the blood, facilitates the binding of the substance to lysine residues present in proteins, or is selected in place of methionine by methioninyl RNA synthetase during protein synthesis.
This process results in the formation of homocysteine-thiolactone in the human body.
It is a chemically reactive thioester metabolite that modifies protein lysine residues in a process called N-homocysteinylation. This modification causes protein damage/aggregation, a hallmark of many diseases. N-homocysteinylated proteins lose their normal function, are cytotoxic, promote immune, inflammatory, thrombotic, and atherogenic processes, and have been linked to cardiovascular disease, Alzheimer's disease, diabetes, kidney disease, neural tube defects, and male infertility.
For this reason, hyperhomocysteinemia is considered a primary risk factor for cardiovascular and other pathogenic events.
The primary pathogenic effects induced by hyperhomocysteinemia are found on the endothelium [35].
The endothelium is the functional unit of the cardiovascular system and is made up of epithelial cells that line the inside of all blood vessels.
The normal endothelium functions as a semipermeable cell layer and acts as a sensor and transmitter of signals in the circulating environment. Endothelial cells are important for maintaining the homeostatic balance of vessels by producing factors that regulate intracellular signaling, vascular tone, coagulation, neutrophil recruitment, lipid transport, hormone trafficking, and the proliferative cellular response, among others.
The pathogenic effect of homocysteine-thiolactone is expressed through the alteration of the proteins that make up the endothelial scaffolding, which produces the breaking of the bonds between the endothelial cells and consequent alteration of the homeostatic functions Fig.15.
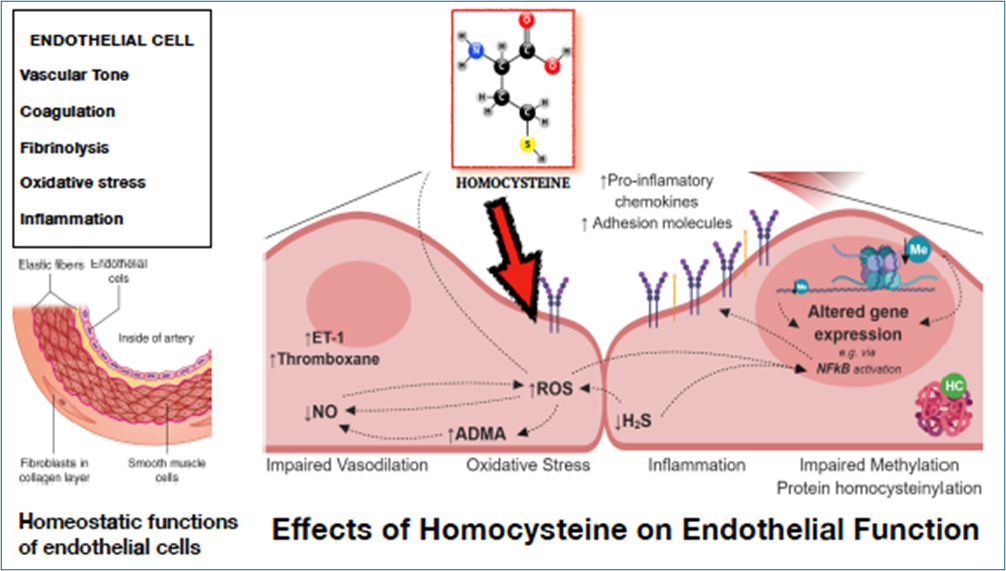
Figure 15- Schematic of the Pathogenic Activity of Homocysteine
Advanced glycation end-products (AGEs
Another class of compounds that are implicated in protein misfolding and conformational changes of other metabolic components are carbohydrates, which are therefore also considered pathogenic causes of many diseases [36-38].
The products thus modified are called advanced glycation end-products (AGEs ) or glycated substances, and are the result of exposure to high concentrations of sugar. The mechanism is called glycation.
Their presence is a pathognomonic index and marker of many degenerative diseases, such as diabetes, atherosclerosis, renal failure, Alzheimer's, etc.
Advanced glycation products are the result of a spontaneous mechanism that occurs between reducing sugars (a reducing sugar is any sugar that, in solution, has an aldehyde or ketone group that acts as a reducing agent) and the autoxidation derivatives with amino groups of proteins, DNA, or lipids, which occur under conditions of hyperglycemia. The formation of AGEs produces negative effects on the structures of macromolecules and determines pathological events.
Their tendency to accumulate takes on a very important pathogenic significance in molecular interactions and can determine macromolecular misfolding similar to that described for proteins.
As an example, we can point out glycated hemoglobin, which is a significant marker of the glycemic status of a diabetic patient.
The glycosylation of macromolecules takes on different meanings based on the group of carbohydrates involved in the process.
Glycans attached to proteins by enzymatic glycosylation can be classified into two types:
- N-glycans, which are those attached to the amide side chain of Asn residues, and
- O-glycans, including those attached to the hydroxyl group of the side chains of Ser or Thr residues.
Cholesterol
Excess cholesterol in the blood is considered a major cardiovascular risk factor, and all primary prevention guidelines recommend keeping it under close monitoring and maintaining its blood concentration below normal values.
Little attention is paid to the fundamental role this substance plays in stabilizing cell membranes, and it represents the most common component within the lipid bilayer of the cell wall.
It contributes to the stability, fluidity, and resistance of the cell membrane, as well as having an important function in modulating the activity of most wall proteins [39-41].
And it is thanks to the association of cholesterol with membrane proteins that the latter function properly. This indissoluble synergistic association between cholesterol and proteins is present in all living organisms and has evolved to perform the functions of the proteins mentioned above.
Until now, only the negative aspects of cholesterol as a potential risk factor have been studied, but the positive role it plays has been overlooked.
Several studies on the cholesterol-protein association have allowed us to examine the association in detail, both from a structural-morphological and functional point of view, and have allowed us to recognize and assign a fundamental role to cholesterol as an essential element for the correct functioning of proteins.
Conclusion
From what we have previously explained, some considerations emerge on the evolution, structural, and functional role of proteins, which makes these substances the fundamental biological component of all vital functions.
Proteins are the result of molecular evolution lasting millions of years, which has produced complex polymeric molecules with the unique ability to change their shape to adapt to the needs imposed by environmental conditions.
Some of them, once they have obtained the optimal size and shape, are able to reproduce and act as a template for forming other proteins of the same shape and size.
Examples of these are prions and amyloid proteins.
These remind us of the behavior of crystals in the formation of crystalline structures.
For this reason, they have played a fundamental role in biological evolution.
This ability to reproduce is a necessary but not sufficient condition for the formation of the extraordinary life forms known to us. Another molecular mechanism was needed to orchestrate the random reproduction of proteins, resulting in autonomous structures designed for reproduction and the necessary adaptations imposed by environmental conditions.
The necessary evolutionary step was the formation of RNA, capable of assembling functionally effective protein forms to carry out useful activities.
The symbiotic relationship between the two RNA-Protein molecular entities has proven to be effective and has been preserved for millions of years.
This system lasted until the revolution with DNA, a molecule capable of collecting and storing environmental information and translating it into phenotypes aimed at reproduction for survival.
To draw an analogy with a system that is more familiar and in continuous use to us, we could compare proteins to writing and language.
Here too, the building blocks are the letters of the alphabet, which consist of 21 characters, at least in Indo-European languages, arranged in succession. Each sentence is like a protein, which must follow logical rules to make sense. An incorrect arrangement of the letters leads to a lack of meaning, just as happens with proteins.
Just as happens with proteins, with the letters of the alphabet, or with the sounds of individual phonemes, it is possible to compose an infinite number of messages, which are exemplified in this article.
Even the gaps between one word and another find analogy in protein synthesis, during which the genetic codes of proteins are separated by stretches of DNA that are not transcribed, and are called introns.
But the analogy doesn't end there. Both in words and with proteins, the messages and forms they express in nature leave us amazed by their beauty and the multiplicity of forms and contents they can convey, making us feel alive and part of a wonderful, complex, and exciting experience, which, in this case, we cannot express in words.
- Wikipedia: Protein structure.
- Elliott J Stollar and David P Smith. (2020). Uncovering protein structure, Essays in Biochemistry. 64(4): 649–680.
- AlphaFold Protein Structure Database Developed by Google DeepMind and EMBL-EBI.
- John Jumper. (2021). Highly accurate protein structure prediction with AlphaFold, Nature. 596(7873): 583-589.
- Aminoacid, Wikipedia.
- Sourav Pan; Amino Acids. (2024). Physical Properties, Structure, Classification, Functions, Biology on Line.
- Deepa Srivastava; Amino Acids, Classification, Chemical Nature, Physical Properties, DDU Gorakhpur University.
- Uversky VN. (2019). Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Front. Phys. 7:10.
- Trivedi, R, Nagarajaram, H.A. (2022). Intrinsically Disordered Proteins: An Overview. Int. J. Mol. Sci. 23(22): 14050.
- Francesco Pesce. (2024). Design of intrinsically disordered protein variants with diverse structural properties, Sci. Adv. 10(35): 9926.
- Mohammad Rehan Ajmal. (2023). Protein Misfolding and Aggregation in Proteinopathies: Causes, Mechanism and Cellular Response, Diseases. 11(1): 30.
- Reynaud, E. (2010). Protein Misfolding and Degenerative Diseases. Nature Education. 3(9): 28.
- F. Ulrich Hartl. (2017). Protein Misfolding Diseases, Annu. Rev. Biochem. 86: 21–26.
- Eszter Herczenik. (2007). The biology of non-native proteins, Printed by Gildeprint B.V, Enschede,The Netherlands.
- Asra Nasir Khan. (2022). Protein misfolding and related human diseases: A comprehensive review of toxicity, proteins involved, and current therapeutic strategies. 223(A): 143-160.
- Michael D. (2015). Geschwind; Prion Diseases, NEUROINFECTIOUS DISEASE. 1612–1638.
- Diane L. Ritchie and Marcelo A. Barria. (2021). Prion Diseases: A Unique Transmissible Agent or a Model for Neurodegenerative Diseases? Biomolecules.11(2): 207.
- L. Kupfer. (2009). Prion Protein Misfolding, Current Molecular Medicine. 9: 826-835.
- Irina L. Derkatch and Susan W. Liebman. (2007). Review, Prion-Prion Interactions, Prion. 1(3): 161-169.
- Alvin S. Das, Wen-Quan Zou. Prions: Beyond a Single Protein, Clin Microbiol Rev. 29(3): 633– 658.
- Yonas I. Tekle and Jessica R. Williams. (2016). Cytoskeletal architecture and its evolutionary significance in amoeboid eukaryotes and their mode of locomotion, R. Soc. open sci. 3: 160283.
- Tim Hohmann and Faramarz Dehghani. (2019). The Cytoskeleton—A Complex Interacting Meshwork, Cells. 8(4): 362.
- Kazumasa Ohashi. (2017). Roles of the cytoskeleton, cell adhesion and rho signalling in mechanosensing and mechanotransduction, J. Biochem.161(3): 245–254.
- Wikipedia; Cytoskeleton.
- Gustavo egea. (2015). Cytoskeleton and Golgi-apparatus interactions: a two-way road of function and structure, Cell Health and Cytoskeleton.
- Jennifer A. Johnston. (1998). Aggresomes: A Cellular Response to Misfolded Proteins, The Journal of Cell Biology. 143(7): 1883–1898.
- Justin T. Marinko. (2019). Folding and Misfolding of Human Membrane Proteins in Health and Disease: From Single Molecules to Cellular Proteostasis, Chem. Rev. 119(9): 5537−5606.
- CT McMurray. (2000). Neurodegeneration: diseases of the cytoskeleton? Cell Death and Differentiation, Macmillan Publishers Ltd. 7(10): 861-865.
- Wikipedia; Protein biosynthesis.
- Cedric Orelle. (2015). Protein synthesis by ribosomes with tethered subunits. Nature. 524(7563): 119–124.
- Laetitia Coassolo. (2025). Understanding peptide hormones: from precursor proteins to bioactive molecules, Trends in Biochemical Sciences. 50(6): 481-494.
- Darren J. Michael. (2006). Mechanisms of peptide hormone secretion, trends Endocrinol Metab. 17(10): 408-415.
- Kuntal Pal, Karsten Melcher, H Eric Xu. (2012). Structure and mechanism for recognition of peptide hormones by Class B G-protein-coupled receptors, Acta Pharmacologica Sinica. 33(3): 300–311.
- Hieronim Jakubowski. (2014). Homocysteine in Protein Structure/Function and Human Disease: Chemical. Biology of Homocysteine-containing Proteins, Springer.
- RC Austin. (2004). Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease, Cell Death and Differentiation. Nature Publishing Group. 11(1):56-64.
- Ana Belén Uceda. (2024). An overview on glycation: molecular mechanisms, impact on proteins, pathogenesis, and inhibition, Biophysical Reviews. 16(2): 189–218.
- Wikipedia; Advanced glycation end-product.
- M. Takahashi. (2007). Glycation and Disease, Comprehensive Glycoscience From Chemistry to Systems Biology. 4: 515-532.
- Muhammad Ahmed. The Role of Cholesterol in Membrane Fluidity and Stability, J Membr Sci Technol. 14(4): 1000408.
- Julie Grouleff. (2015). The influence of cholesterol on membrane protein structure, function, and dynamics studied by molecular dynamics simulations, Biochimica et Biophysica Acta. 1848 (2015): 1783–1795.
- Matthias Pöhnl. (2023). Nonuniversal impact of cholesterol on membranes mobility, curvature sensing, and elasticity, Nature Communications. 14: 8038.
Download Provisional PDF Here
PDF