

p (1).png)



.png)




.png)

Research Article
Structure-Based Screening and Molecular Dynamics Simulation Studies for the Identification of Potential Gsk-3β Inhibitors
- Dr. Ashish Sarkar
Corresponding author: Dr. Ashish Sarkar, Dean School of Pharmacy YBN University Ranchi- Jharkhand-834010.
Volume: 2
Issue: 3
Article Information
Article Type : Research Article
Citation : Shouvik Mal, Dr. Ashish Sarkar. Structure-Based Screening and Molecular Dynamics Simulation Studies for the Identification of Potential Gsk-3β Inhibitors. Journal of Medicine Care and Health Review 2(3). https://doi.org/10.61615/JMCHR/2025/OCT027141004
Copyright: © 2025 Dr. Ashish Sarkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
DOI: https://doi.org/10.61615/JMCHR/2025/OCT027141004
Publication History
Received Date
15 Sep ,2025
Accepted Date
30 Sep ,2025
Published Date
04 Oct ,2025
Abstract
The goal of researchers for almost twenty years has been to discover ways to directly block glycogen synthase kinase 3β (GSK3Ų). Structure-based virtual screening (SBVS) and high-throughput enzyme assays were used in order to search through a library of 3,000 compounds in order to identify potential novel GSK3β inhibitors. In comparison to the other compounds, the pyrazolo[1,5-a]pyrimidin-7-amine derivatives stood out owing to their powerful inhibitory action on GSK3β and their noteworthy modification of Wnt signaling pathways. The subsequent SBVS was directed by molecular dynamics (MD) simulations that were supplemented with upper-wall limitations. These simulations assisted in the refinement of the structural understanding of ligand binding. Through the use of this integrated strategy, the identification of highly effective and selective GSK3β inhibitors, as well as the acquisition of extensive knowledge on their binding mechanisms, became feasible. The findings of this research have the potential to provide the foundation for the enhancement of potential therapeutic medicines that target GSK3β within the hunt for viable therapeutic agents.
Keywords: GSK-3β inhibitors, Structure-based virtual screening (SBVS), Molecular dynamics (MD) simulation, Enzyme inhibition
►Structure-Based Screening and Molecular Dynamics Simulation Studies for the Identification of Potential Gsk-3β Inhibitors
Shouvik Mal1, Dr. Ashish Sarkar2*
1PG Scholar School of Pharmacy YBN University.
2Dean School of Pharmacy YBN University Ranchi- Jharkhand-834010.
Introduction
The serine/threonine kinase GSK-3β is associated with several physiological processes, including the control of cell cycle progression, glycogen metabolism, and nervous system function [1]. GSK-3β activity issues have been associated with a variety of illnesses, such as cancer, bipolar disorder, and Alzheimer's disease, among others. As a result, GSK-3β has become a well-recognized therapeutic target.
Challenges in Developing GSK-3β Inhibitors
Even though there has been a significant amount of research conducted on the issue, it has been challenging to develop GSK-3β inhibitors that are both selective and potent. Because of the presence of a multitude of phosphorylation sites and a highly conserved ATP-binding pocket, it is challenging to discover inhibitors that are specifically designed to target this kinase [2]. In addition, achieving adequate penetration of the blood-brain barrier is a significant obstacle for applications involving the central nervous system.
Structure-Based Virtual Screening (SBVS) in Drug Discovery
There are several physiological processes that are linked to the serine/threonine kinase GSK-3β. These activities include the regulation of the progression of the cell cycle, the metabolism of glycogen, and the functioning of the nervous system [3]. This method is referred to as structure-based virtual screening. In order to determine which compounds should be prioritized for further experimental validation, SBVS docks a library of compounds into the binding site of the target and then predicts the binding affinity and selectivity of those compounds.
Role of Molecular Dynamics (MD) Simulations
Simulations of molecular dynamics (MD) give an atomic-level insight into the interactions that occur between proteins and ligands over the course of time [4]. When it comes to GSK-3γ, molecular dynamics (MD) simulations have the ability to explain how the enzyme-inhibitor complex navigates through time, identify key residues that are involved in binding, and forecast how the complex will behave when subjected to physiological loading.
Integration of SBVS and MD Simulations
There is an improvement in the drug development process when SBVS is linked with MD simulations [5]. This improvement is achieved by combining the effectiveness of virtual screening with the precision of dynamic simulations. With the assistance of this all-encompassing technology, which discovers GSK-3β inhibitors with high affinity and provides information on their binding mechanisms, it is possible to build a therapeutic medicine that is both more potent and selective.
Objectives
- To use structure-based virtual screening (SBVS) to find possible GSK-3β inhibitors from a chemical library.
- To evaluate the stability and binding interactions of certain inhibitors using molecular dynamics (MD) simulations.
Materials And Methods
Simulation of Molecular Docking
The crystal structure of GSK3β with a 2.4 Å resolution (PDB ID: 1PYX) was the starting point for the molecular modeling investigation [7]. Using the MODELER auto model and loop model in Discovery Studio (DS) 2018 [8], the missing loop sections of the crystal structure were created. The protein was prepared for molecular docking by adding hydrogen atoms and then purging it of any ligands and water. The GSK3β active site was located within 12 Å of AMP-PNP, an insoluble analogue of ATP. In order to build SB216763 and achieve compound starting docking postures, this molecular dynamics (MD) investigation used CDOCKER [9], a docking simulation program in DS 2018. All hit derivatives were compared using Schrödinger's Glide (version 2019-1, Schrödinger, LLC: New York, NY, USA, 2017).
Simulation of Molecular Dynamics (MD)
We ran all-atom MD simulations on the best docking postures from GROMACS 2016 using the Plumed plugin, version 2.4 [10]. The TIP3P explicit solvent model was used, with the ligand and protein modeled using the charmm36 all-atom force field. With the help of CHARMM-GUI, the simulation input was generated [11,12]. The CHARMM General Force Field (CGenFF) approach was used to build the ligand topologies and parameters [13]. A solvated cubic water box that is 10 Å thick is present in every system. A solution of 0.15 M NaCl was used to neutralize the system. In contrast to the cpd1 system, which had 70 Na+ and 77 Cl−, the SB216763 had 61 Na+ and 68 Cl−. System energy minimization via steepest descent was continued until a tolerance of 1000 kJ/mol was obtained in order to eliminate faulty connections. The reduced structures underwent NVT equilibration at 303.15 K with a 1 fs time step for 25 ps, where CPN, VVT, and T are constants. Hydrogen atom couplings could only be as long as the LINCS algorithm determined their equilibrium bond length [14]. The Nosé-Hoover thermostat [15,16] and the Parrinello-Rahman barostat [17] were used to conduct NPT dynamics over four 500 ns manufacturing cycles. Out of the four runs, three were performed on SB216763, and one on cpd1. We kept the temperature at 303.15K. The trajectory was stored every picosecond, and the time step was set to 2 fs for the production runs. At 12 Å, the cut-off values for short-range electrostatic interactions and Van der Waals were determined. For long-distance electrostatic interactions, the particle mesh Ewald method [18] was used. It was common practice to take a picture from the second trajectory, which had the lowest non-bond energy between the ligand and protein, and a highly packed conformation over the final 200 ns, in order to analyze protein-ligand interactions. When the distance between the residues at the binding site and the ligand COM exceeded 12 Å, an upper-wall restraining force was applied to stop the ligand from escaping to the bulk solvent. According to the formula κ = 200 kJ/mol nm−2, the wall's harmonic potential was calculated. Constraint force was determined using the following equation.
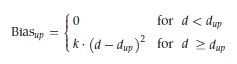
All of the results from the simulation analyses were obtained using Visual Molecular Dynamics (VMD), version 1.9.4a12 [19], GROMACS 2016, and DS 2018.
Pharmacophore Model Creation and Virtual Screening
The most crucial pharmacophoric characteristics for inhibitor binding were derived from the typical structure of SB216763's 455.8 ns snapshot. The data was analyzed using DS 2018's Interaction Pharmacophore Generation. By choosing four or six features using the receptor-ligand pharmacophore generating approach, we created a realistic-sized model. Based on protein-ligand non-bond interactions, this technique builds pharmacophore models with specified features. The created pharmacophore model was coupled with the Search 3D Database approach in DS 2018 to retrieve active chemicals from the Korea Chemical Bank (KCB)'s 530,000 compounds. This search utilized the KCB database, a DS 2018 Build 3D Database protocol-built indexed multi-conformer database. The fit value, clustering, and patent filtering determined the final three thousand compounds to be evaluated in vitro.
GSK3β Kinase Assay High-Throughput Screening (HTS) (TR-FRET)
At 25 °C, 384-well white plates (Greiner) and a Biomek FX system (Beckman Coulter) were used to perform in vitro enzyme TR-FRET assays. Ten microliters of kinase buffer containing 50 millimolar HEPES (pH 7.5), 1 millimolar EGTA, 10 millimolar MgCl₂, 2 millimolar DTT, and 0.01% Tween-20 were used in the experiment. A 5 mM DMSO stock was used to select 3,000 compounds for virtual screening. These compounds were then properly diluted with the kinase buffer. To make the assay plates, 5 µL of GSK3β (0.5 ng/well, Carna Biosciences) and 2.5 µL of the compounds were added. Then, 2.5 µL of ULight-GS Peptide (Ser641/pSer657, PASVPPSPSLSRHSSPHQ(pS)ED, #TRF0131 PerkinElmer, 50nM final concentration) and ATP were added. At the end of the first hour, 10 µL of a stop solution containing 24 mM EDTA and 2 nM of Eu-anti-phospho-GS (Ser641) antibody was added to end the reaction, which was then incubated for at least another hour. The Envision Multilabel Plate Reader (PerkinElmer) was used to measure the fluorescence signals at 320 nm excitation and 665 nm emission. GraphPad Prism 7.0 with nonlinear regression was used to determine IC₅₀ values from dose-response curves. Lineweaver-Burk plots were used to investigate competitive inhibition. A five-by-five matrix of ATP and inhibitor concentrations was used for this analysis. The following steps were used in order to conduct reversibility studies: first, GSK3β (1 ng/µL, 20 µL) was pre-incubated with compounds that were 20 times concentrated for 30 minutes. Then, 6.5 µL of kinase buffer and 1 µL of GSK3β/compound combination were added, followed by 2.5 µL of Peptide/ATP mixture. The results are shown as the mean ± SD (n = 2), and the reactions were halted at various times in time. All of the high-throughput screening tests used SB216763 as a reference GSK3β inhibitor.
Assay for Reversibility
Twenty microliters of GSK3β (1 ng/µL) and twenty microliters of the compound that was twenty times concentrated were pre-incubated for thirty minutes. The well was supplemented with 6.5 µL of kinase buffer and 1 µL of the GSK3β/compound combination. Shortly after, 2.5 µL of the Peptide/ATP combination was added. Different incubation durations were found to halt the process.
Assay for Reporters
The TOPflash-inserted HEK293 reporter cells were generously donated by KY Choi, a researcher from Yonsei University in Seoul, Korea. Reporter cells from HEK293 were grown in Gibco's DMEM supplemented with 10% FBS. A 96-well black polystyrene plate (Greiner Bio-One, Kremsmünster, Austria) was used to plate HEK293 reporter cells, 1.5 x 10¹ cells per well, in order to assess the effect of GSK3β inhibition on hit compounds. In a day, the cells multiply. One or more of the following was added to each well: hit compounds, DMSO, or a positive control (CHIR99021, SelleckChem, Houston, TX, USA). After 18 hours, the firefly luciferase activity of the plates was assessed, and the relative reporter activity was measured after comparing it to the cell viability study of the WST-1 (Takara Korea Biomedical Inc., Seoul, Korea). For the statistical analysis, GraphPad Prism 8 was used. We used a one-way ANOVA test with significance levels of * p < 0.05, ** p < 0.005, and *** p < 0.001 to examine the impact of each group.
Result And Discussion
Reference Compound SB216763: Molecular Dynamics (MD) Simulation Using GSK3β
Successful structure-based virtual screening required a robust protein-ligand pharmacophore model for filtering. This model would not have been possible without the adjusted binding site structure to the extremely active chemical. The SB216763 compound, with an IC50 value of 34 nM [20], was used by docking at the ATP-binding site of GSK3γ (PDB ID: 1PYX). AMP-PNP was removed from the crystal structure to find novel scaffolds that did not replicate ATP-binding. SB216763 was initially discovered via molecular docking. The redesigned protein-ligand complex structure was then simulated using MD.
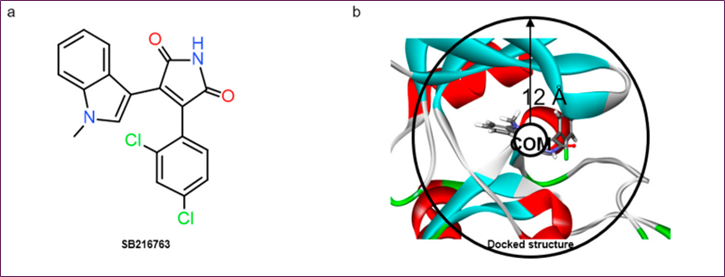
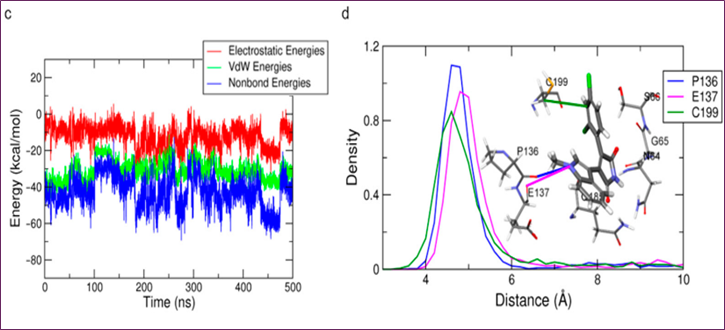
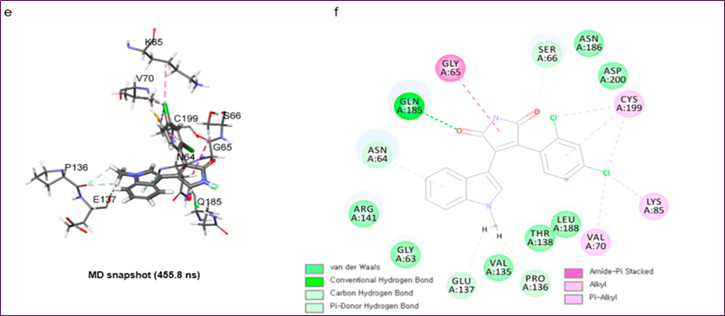
Figure 1. MD simulation analysis of SB216763 bound to GSK-3β: (a) chemical structure, (b) upper-wall restraint, (c) protein–ligand non-bonded energies, (d) distance distribution with key residues, (e) 3D binding mode, (f) 2D interaction diagram
In comparison to molecular docking, the protein-ligand flexibility was enhanced by solvent effects in the MD simulation, leading to better non-bonded interactions and structural modifications. All of these benefits are rendered useless since it takes several hundred nanoseconds for the ligand to return to the binding site. Since there is a much smaller unbinding barrier from the binding site to the solvent region compared to the binding barrier, the ligand is more likely to be in the solvent area. In order to effectively determine a binding mode when the distance between the residues at the binding site and the ligand SB216763 was more than 12 Å, the harmonic potential (κ = 200 kJ/mol nm−2) was applied around the binding site using a constraint force (Figure 1b). If the COM-ligand distance is more than 12 Å, then apply this force only when the ligand is prepared to leave the binding site.
Three separate 500 ns MD simulations were performed using the same parameters in order to get the structure of the protein-ligand complex. The RMSD study showed that all three trial systems had well-stabilized systems and ligands at 3~5 Å and 0.75~1.25 Å, respectively. In the first two trials, SB216763's binding mode was consistent; however, in the third trial, it changed. The second trial trajectory yielded the three-dimensional snapshot at 455.8 ns, which is the most common binding mode and has the lowest protein-ligand energy (Figure 1c). In the final 200 nanoseconds, the distribution of distances between critical residues was determined for the binding mode analysis of SB216763 with GSK3β protein (Figure 1d). Residues G65, S66, and Q185 in the loop region lost their hydrophobic fit to the ATP-binding site, the Pi-alkyl contact between C199 and the ligand, and the carbon-hydrogen link between SB216763 and the backbone oxygen of P136 and E137. View Figures 1e and f. The binding mode was stable during the last 200 ns of the experiment.
Screening Virtually
Structure-based virtual screens, docking, and MD simulations were used to identify new GSK3β inhibitors in Korea Chemical Bank (KCB) libraries (Figure 2a). To build a typical protein-ligand interaction structure, SB216763 was docked with the GSK3β protein's ATP-binding domain and simulated using MD software. As said, the 455.8 ns snapshot was selected since it had the most binding modes with favorable non-bond energies.
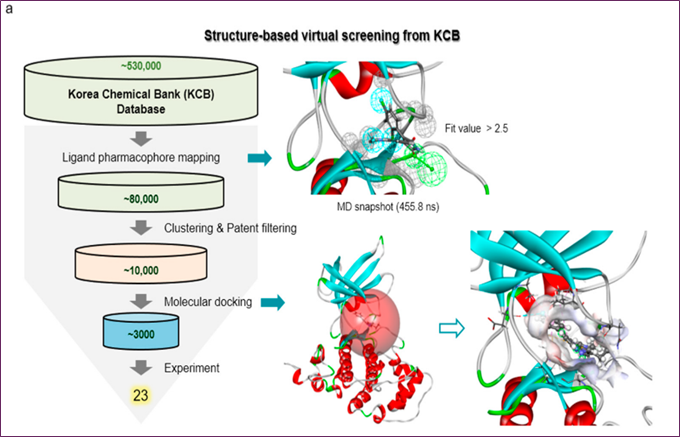
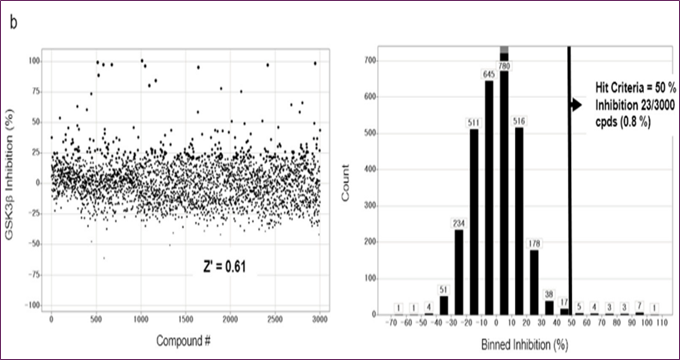
Figure 2. Methods for finding GSK-3β inhibitors via the use of molecular docking, structure-based virtual screening, and MD simulations: (a) The impact compound cpd1-using SBVS method, (b) the distribution of activities
One hydrogen bond acceptor and three hydrophobic ones make up this structure, which is the basis of the three-dimensional protein-ligand pharmacophore model. The KCB database, which contains 530,000 compounds, was searched for chemicals using a 3D query. By using a fit value higher than 2.5, 80,000 compounds were filtered out. After screening for patents and considering chemical variety, almost 10,000 compounds were selected. After the compounds were modeled using molecular docking software, the top three thousand molecules were subjected to an in vitro test.
Enzyme Assays in Vitro and the Subsequent HTS Campaign
Compounds with GSK3β inhibitory activities were tested using a Lance HTRF assay. The Z factor, 0.61, and S/N ratio were obtained at 10 μM, whereas the Z' factor was 0.48 and the Z factor was 7.3. Activity distribution for the high-throughput screening effort is shown in Figure 2b. After choosing 23 compounds (0.8%) with a 50% hit criterion, we calculated the IC50 values using dose-response testing. After visually picking one of the 23 hit molecules, we searched KCB for similar compounds. Nearly 100 compounds were identified using KCB IC50 values. Finally, five candidate drugs were discovered (Table 1), including cpd1, which struck at 40 nM (Figure 3a). Figure 3b demonstrates competitive suppression of GSK3β-catalyzed phosphorylation by cpd1, as determined by a five-by-five-concentration kinetic study. The slope inhibition constant K is 41.8 nM. Competitive inhibition studies may reveal inhibitor behaviour and enzyme binding potency. Medicines' effects on enzymes are characterized by their IC50 values. Several drugs compete with the target enzyme. However, substrate concentration affects IC50 values, especially for competitive inhibitors. Thus, enzyme kinetic studies favor Ki values based on the linear reaction period's starting velocity. It seemed that Cpd1 and ATP fought for the same binding site. We then examined cpd1's enzyme-binding kinetics using dilution. ATP-competitive and reversible, cpd1 is like SB216763, the reference competitive inhibitor (Figure 3c).
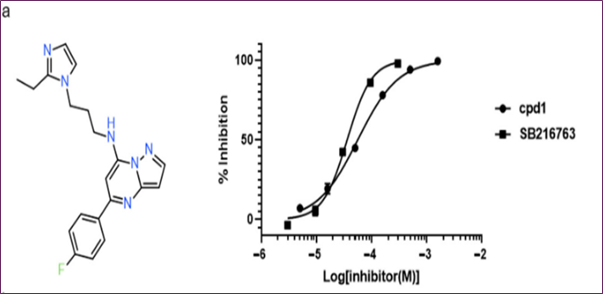
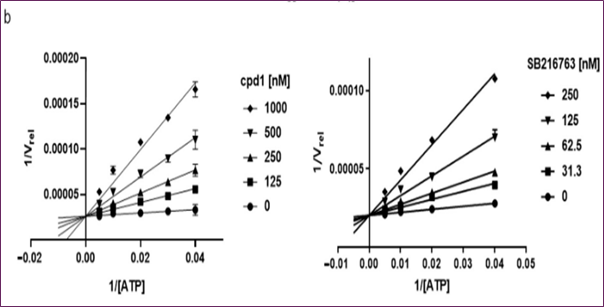
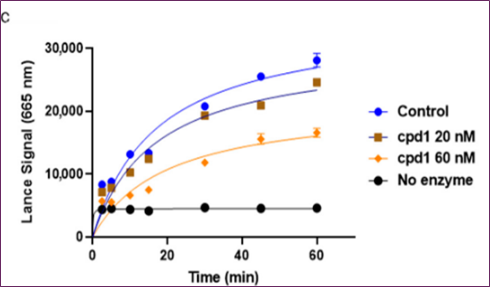
Figure 3. In vitro evaluation of cpd1: (a) structure and dose–response curves with SB216763, (b) competitive inhibition at varying ATP concentrations, (c) reversibility test.
Table 1. Effects of pyrazolo derivatives on GSK3β inhibition [1,5-a] Pyrimidine.
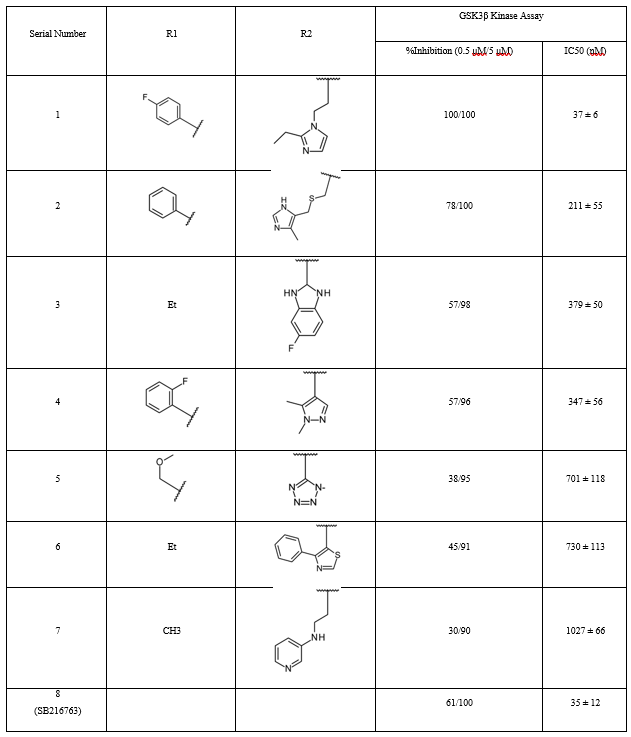
Cpd1 Hit Compound Binding Mode Analyses
Molecular docking and MD simulations of cpd1 were conducted using the protein structure to determine the most effective drug binding mechanism. If you want to run simulations, you need a 455.8 ns snapshot. The first cpd1 docking posture was identified by the CDOCKER molecular docking method. After running a 500 ns MD simulation on cpd1, the docking location was fine-tuned. At 3 Å and 2 Å, respectively, the RMSD study showed that the system and ligand were well-stabilized. In order to analyze the mechanism of cpd1 binding, the distribution of distances between important residues during the last 200 ns was computed (Figure 4a). Hydrogen connections were formed between the oxygen in Q185's backbone and cpd1. I62, V70, A83, Y140, L188, and C199 were shown to interact with Pi-alkyl groups and hydrophobic groups in Figure 4c,d. The loop N64 link was not as stable as the hydrogen bond connection that the Q185 backbone oxygen maintained with the hit molecule for 200 ns.
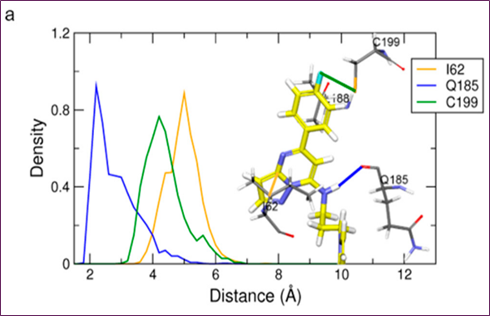
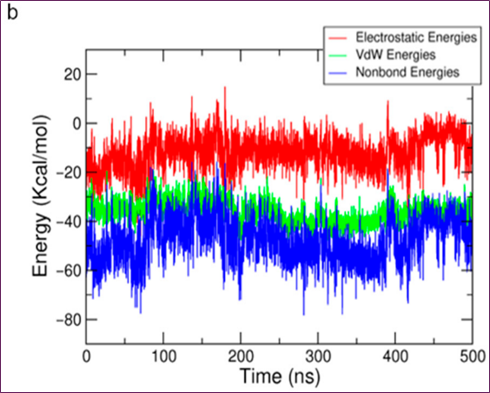
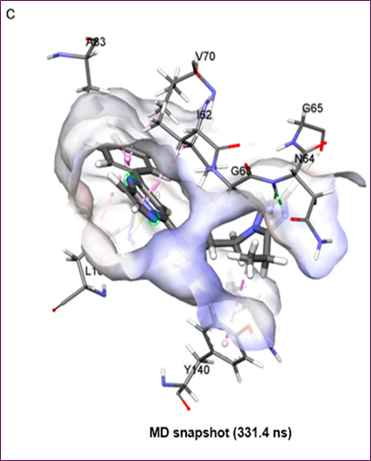
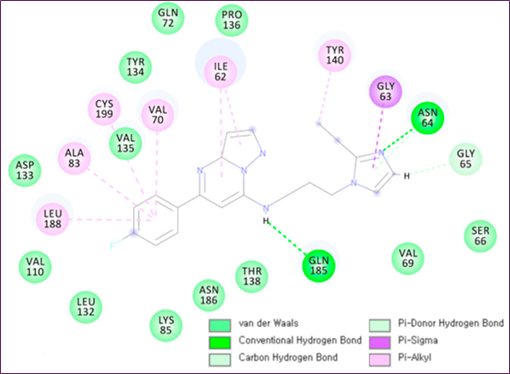
Figure 4. MD simulation analysis of hit compound cpd1: (a) distance distribution with key residues, (b) protein–ligand non-bonded energy, (c) 3D binding mode, (d) 2D interaction diagram
Despite cpd1's ethyl-imidazole moiety undergoing substantial simulational deviance, the core scaffold of pyrazolo[1,5-a]pyrimidine-7-amine with fluorobenzene was stable for 200 ns. Figure 4b,c reveals that the 331.4 ns snapshot had the usual cpd1 structure, which had the lowest protein-ligand energy and was densely packed. Compared to the reference compound and final representative structure, cpd1's non-bond energy (−77.95 kcal/mol) was comparable to SB216763's (−68.53 kcal/mol).
A Molecular Docking Study of Hit Derivatives
Molecular docking simulations were performed using CDOCKER and Glide with the typical cpd1 structure at 331.4 ns to examine the binding modalities of the extra hit modifications. Because the Glide energy score was most strongly correlated with docking scores (r = 0.80) in the IC50 test (Table 2), we used the final docked postures from the Glide docking simulation to compare the poses. The docked position of cpd1 was almost equal to its location in the usual structure at 331.4 ns, with an RMSD of 1.08 Å. As we evaluated the core scaffold alignment across derivative docked conformations, Figure 5 demonstrates a good linear connection (r = 0.98) between Glide energy values and experimental IC50 values (Table 2). The IC50 of five active compounds was less than 1 μM, indicating good alignment.
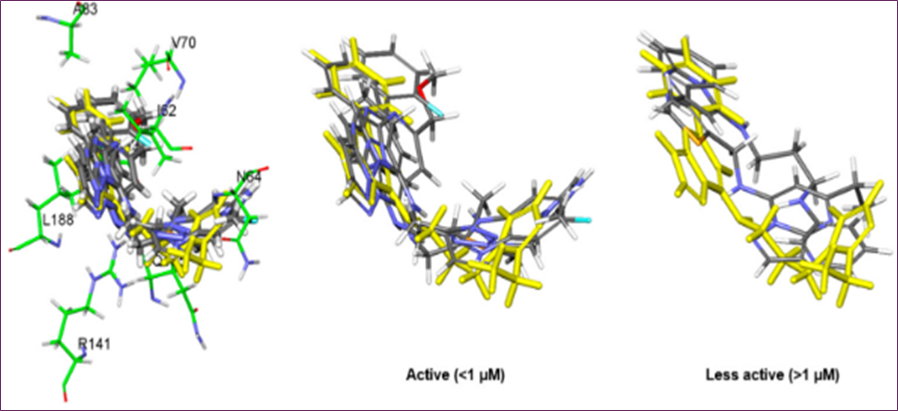
Figure 5. Molecular docking of hit derivatives: overlap of five highly active compounds (IC₅₀ < 1 µM) and two less active compounds, highlighting cpd1
Table 2. Molecular docking results of hit derivatives: Glide binding energy, negative CDOCKER energy (CE), and interaction energy (CIE)
|
Compound |
IC50 (nM) |
Glide_Evdw |
Glide_Ecoul |
Glide_Energy |
Glide_XP_Hbond |
CIE |
CE |
|
1 |
40 |
−43.94 |
−7.05 |
−50.99 |
−1.08 |
45.67 |
21.48 |
|
2 |
254 |
−40.66 |
−7.07 |
−47.73 |
−0.35 |
41.11 |
10.56 |
|
3 |
334 |
−33.39 |
−7.56 |
−40.95 |
−0.97 |
33.25 |
8.16 |
|
4 |
485 |
−39.62 |
−2.47 |
−42.09 |
−0.31 |
34.49 |
−56.24 |
|
5 |
776 |
−30.95 |
−4.09 |
−35.04 |
−0.40 |
39.27 |
−3.75 |
|
6 |
1166 |
−40.90 |
−2.14 |
−43.04 |
−0.03 |
34.04 |
10.12 |
|
7 |
1190 |
−33.86 |
−3.85 |
−37.71 |
0 |
33.13 |
13.95 |
Activity of Wnt Signaling in Relation to the Active Ingredients
Stabilizing β-catenin and activating Wnt/β-catenin signaling might be achieved by pharmaceutical suppression of GSK3β activity. To determine the inhibitory effects of compounds, HEK293 TOP flash reporter cells were examined at different doses in a GSK3β enzyme-based high-throughput screening experiment. Out of all the hits, cpd1 demonstrated a notable rise in TOP flash activity, with a dose-dependent induction that was 3.5 times higher at 5 µM (Figure 6).
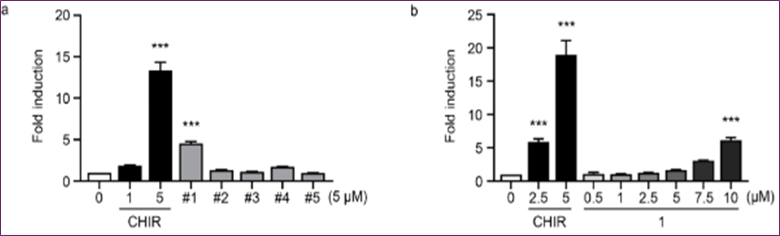
Figure 6. The TOP flash reporter experiment in HEK293 cells and the EC₅₀ measurement for cpd1 show that cpd1 activates Wnt signaling.
Without stimulation, GSK3β is constantly active and expressed in all cells. Autophosphorylation of Tyr216 or mediation by PYK2, MEK1, or SRC kinases positively regulates it, whereas AKT phosphorylates it on Ser9 negatively [21,22]. When activated, GSK3β plays a complex function in several physiological processes, phosphorylating over 40 proteins, including over 12 transcription factors. GSK signaling regulates brain development's progenitor cell proliferation and differentiation. However, aberrant GSK3β and NF-κB activation is connected to neuroinflammation, epilepsy, and Alzheimer's disease [23]. GSK3β phosphorylates β-catenin in unstimulated (Wnt turned-off) cells to reduce its cellular levels due to eventual ubiquitination/degradation. When GSK3β inhibitors or Wnt bind to its heterodimeric receptor (Frizzled receptor and LRP5/6 co-receptor), β-catenin stabilizes and activates gene transcription. Additionally, GSK3β and CK-1 phosphorylation increase LRP5/6 and Wnt signaling. Although it has many biological consequences, blocking GSK3β stimulates the Wnt/β-catenin and PI3K/PTEN/Akt/mTORC1 pathways. The inhibitors used for Figure 3 demonstrated that blocking the GSK3β enzyme enhanced Wnt signaling in cells. Inhibitors of GSK3β might potentially facilitate the processes of neurogenesis and endoderm differentiation [24]. GSK3β over-activation has been associated with cancer, diabetes, and degenerative brain diseases [25]. Targeting GSK3β has several potential uses. Virtual modeling and high-throughput screening are the most effective methods for identifying a druggable scaffold.
The SBVS-experimental HTS research did have one limitation, though: they only tested the modeling technique with a single target protein. The applicability of this tactic to different objectives remains uncertain. Our prior work [26] and others' [27,28] demonstrate, however, that MD modeling enhances molecular docking outcomes. To improve the precision of the protein-ligand structure and to speed up the process, this work used an upper-wall constraint force in an MD simulation. The last pharmacophore model for computational screening was generated by this process. Our method effectively found inhibitors of GSK3β by drawing on prior research [29,30] and current literature.
Conclusion
Molecular docking and upper-wall limited molecular dynamics (MD) simulations were used in this study to efficiently uncover compounds that have the potential to inhibit GSK-3β. Additionally, a technique for virtual screening compounds based on structures was successful in identifying these compounds. In order to discover potential compounds that target the ATP-binding site, the MD-derived binding properties of the reference molecule SB216763 were used. According to the findings of the molecular modeling research, the pyrazolo[1,5-a]pyrimidine-7-amine scaffold exhibits beneficial binding modes. This is because it establishes a greater number of hydrogen bonds and hydrophobic interactions than the reference. The discovery of five possible hit compounds as novel GSK-3β inhibitors has established a strong foundation for the future optimization of leads and the creation of a potent therapeutic medication.
- Zhou, H, Gao, Y, Li, Y. (2022). Structure-based virtual screening for GSK-3β inhibitors: Insights from molecular docking and dynamics simulations. Journal of Molecular Modeling. 28(4): 100.
- Wang, J, Zhang, Q, Li, X. (2020). Integrating machine learning-based virtual screening with multiple protein structures for discovery of novel GSK3β inhibitors. Frontiers in Pharmacology. 11: 605608.
- Eldar-Finkelman, H, Martinez, A. (2011). Glycogen synthase kinase 3 inhibitors: Preclinical and clinical focus on CNS disorders. Current Drug Targets. 12(7): 892–905.
- Jain, A, Chawla, M. (2017). Molecular docking and dynamics simulations of GSK-3β inhibitors: Insights into binding interactions. Journal of Biomolecular Structure and Dynamics. 35(11): 2330–2341.
- Li, X, Zhu, Q. (2019). Advances in GSK-3β inhibitor design using computational approaches. European Journal of Medicinal Chemistry. 162: 273–289.
- Ali, S, Ahmad, A. (2021). Computational identification of GSK-3β inhibitors via structure-based screening and molecular dynamics simulation. Computational Biology and Chemistry. 95: 107552.
- Bertrand, J. Thieffine, S. Vulpetti, A. Cristiani, C.Valsasina, B. Knapp, S. Kalisz, H. Flocco, M. (2003). Structural Characterization of the Gsk-3β Active Site Using Selective and Non-Selective Atp-Mimetic Inhibitors. J. Mol. Biol. 333(2): 393–407.
- BIOVIA. (2018). Dassault Systèmes. BIOVIA Discovery Studio, Release 2018, San Diego: Dassault Systems.
- Wu, G.Robertson, D.H,Brooks, C.L, Vieth, M. (2003). Detailed Analysis of Grid-Based Molecular Docking: A Case Study of Cdocker—A Charmm-Based Md Docking Algorithm. J. Comput. Chem. 24(13): 1549–1562.
- Tribello, G.A, Bonomi, M, Branduardi, D. Camilloni, C, Bussi, G. (2014). Plumed 2: New Feathers for an Old Bird. Comput. Phys. Commun. 185(2): 604–613.
- Jo, S, Kim, T, Iyer, V.G, Im, W. Charmm-Gui. (2008). A Web-Based Graphical User Interface for Charmm. J. Comput. Chem. 29(11): 1859–1865.
- Lee, J, Cheng, X, Swails, J.M, Yeom, M.S, Eastman, P.K, Lemkul, J, Wei, S, Buckner, J, Jeong, J.C, Qi, Y. (2016). Charmm-Gui Input Generator for Namd, Gromacs, Amber, Openmm, and Charmm/Openmm Simulations Using the Charmm36 Additive Force Field. J. Chem. Theory Comput. 12(1): 405–413.
- Vanommeslaeghe, K, Hatcher, E, Acharya, C, Kundu, S, Zhong, S, Shim, J, Darian, E, Guvench, O, Lopes, P, Vorobyov, I. (2010). Charmm General Force Field: A Force Field for Drug-Like Molecules Compatible with the Charmm All-Atom Additive Biological Force Fields. J. Comput. Chem. 31(4): 671–690.
- Hess, B, Bekker, H, Berendsen, H.J, Fraaije, J.G. Lincs. (1997). A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 18(12): 1463–1472.
- Hoover, W.G. (1985). Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A. 31: 1695.
- Nosé, S. (1984). A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 52(2): 255–268.
- Parrinello, M, Rahman, A. (1981). Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 52: 7182–7190.
- Essmann, U, Perera, L, Berkowitz, M.L, Darden, T, Lee, H, Pedersen, L.G. (1995). A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 103: 8577–8593.
- Humphrey, W, Dalke, A, Schulten, K. (1996). Vmd: Visual Molecular Dynamics. J. Mol. Graph. 14(1): 33–38.
- Coghlan, M.P, Culbert, A.A, Cross, D.A, Corcoran, S.L, Yates, J.W, Pearce, N.J, Rausch, O.L, Murphy, G.J, Carter, P.S, Cox, L.R. (2000). Selective Small Molecule Inhibitors of Glycogen Synthase Kinase-3 Modulate Glycogen Metabolism and Gene Transcription. Chem. Biol. 7(10): 793–803.
- Hartigan, J.A, Xiong, W.-C, Johnson, G.V. (2001). Glycogen Synthase Kinase 3β Is Tyrosine Phosphorylated by Pyk2. Biochem. Biophys. Res. Commun. 284(2): 485–489.
- Hoffmeister, L, Diekmann, M, Brand, K, Huber, R. (2020). Gsk3: A Kinase Balancing Promotion and Resolution of Inflammation. Cells. 9(4): 820.
- Toral-Rios, D, Pichardo-Rojas, P.S, Alonso-Vanegas, M, Campos-Peña, V. (2020). Gsk3β and Tau Protein in Alzheimer’s Disease and Epilepsy. Front. Cell. Neurosci. 14: 19.
- Huang, J, Guo, X, Li, W, Zhang, H. (2017). Activation of Wnt/Β-Catenin Signalling Via Gsk3 Inhibitors Direct Differentiation of Human Adipose Stem Cells into Functional Hepatocytes. Sci. Rep. 7: 40716.
- Valvezan, A.J, Klein, P.S. (2012). Gsk-3 and Wnt Signaling in Neurogenesis and Bipolar Disorder. Front. Mol. Neurosci. 5: 1.
- Lee, Y, Kim, S, Kim, J.Y, Arooj, M, Kim, S, Hwang, S, Kim, B.-W, Park, K.H, Lee, K.W. (2014). Binding Mode Analyses and Pharmacophore Model Development for Stilbene Derivatives as a Novel and Competitive Class of A-Glucosidase Inhibitors. PLoS ONE. 9(1): 85827.
- Salo-Ahen, O.M.H, Alanko, I, Bhadane, R, Bonvin, A.M.J.J, Honorato, R.V, Hossain, S, Juffer, A.H, Kabedev, A, Lahtela-Kakkonen, M, Larsen, A.S. (2020). Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes. 9(1): 71.
- Guterres, H, Im, W. (2020). Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Model. 60(4): 2189–2198.
- Kim, D.G, Choi, Y, Lee, Y, Lim, S, Kong, J, Song, J, Roh, Y, Harmalkar, D.S, Lee, K, Goo, J.-I. (2022). Aimp2-Dx2 Provides Therapeutic Interface to Control Kras-Driven Tumorigenesis. Nat. Commun. 13: 2572.
- Kim, D.G, Huddar, S, Lim, S, Kong, J, Lee, Y, Park, C.M, Lee, S, Suh, Y.-G, Kim, M, Lee, K. (2021). Allosteric Inhibition of the Tumor-Promoting Interaction between Exon 2–Depleted Splice Variant of Aminoacyl–Transfer Rna Synthetase-Interacting Multifunctional Protein 2 and Heat Shock Protein 70. J. Pharmacol. Exp. Ther. 379(3): 358–371.
Download Provisional PDF Here
PDF